EMA Regulatory
Integral DDC Labeling: The CE Mark Trap Nobody Warns You About
Quick Answer
Integral DDC labeling follows pharmaceutical legislation-not MDR labeling requirements. The CE mark and UDI should not appear on the iDDC's outer packaging, even if the device component was previously CE marked. Device Instructions for Use must be integrated into the SmPC and Package Leaflet using QRD templates. The device's administrative information (manufacturer details, NB number) stays off the medicinal product labeling entirely.
Part 3 of a deep-dive series analyzing EMA's Questions and Answers on MDR/IVDR implementation. Part 1 covered Q1: DDC Classification. Part 2 covered Q2: Borderline Product Advice. This post examines Q3-4-why labeling an integral DDC requires unlearning your device labeling instincts.
A colleague forwards him a marked-up draft of the package leaflet. Seven annotations in red, all in the same section-the device instructions.
The comments are from the quality reviewer, and they all say variations of the same thing: this reads like a device IFU, not like a section of a medicinal product leaflet. The formatting is wrong. The symbols are wrong. And there's a CE mark on the mock-up that shouldn't be there at all.

He's been working on medical devices for eight years. He knows MDR Annex I labeling requirements in his sleep-every symbol, every mandatory element, every placement rule. And that's precisely the problem. Because integral DDCs don't follow those rules. They follow pharmaceutical labeling rules, and the instincts he's built over a decade are now working against him.
EMA's Q&A Questions 3 and 4 explain why-and what integral DDC labeling actually requires. The logic is clear once you see it. The challenge is unlearning what you already know about device labeling.
What Q3 and Q4 Actually Address
Why this matters: Q3 and Q4 resolve a tension that exists at the heart of integral DDCs-a product with device components that must follow pharmaceutical labeling rules. Understanding the EMA's position prevents labeling deficiencies that create review cycles and delay authorization.
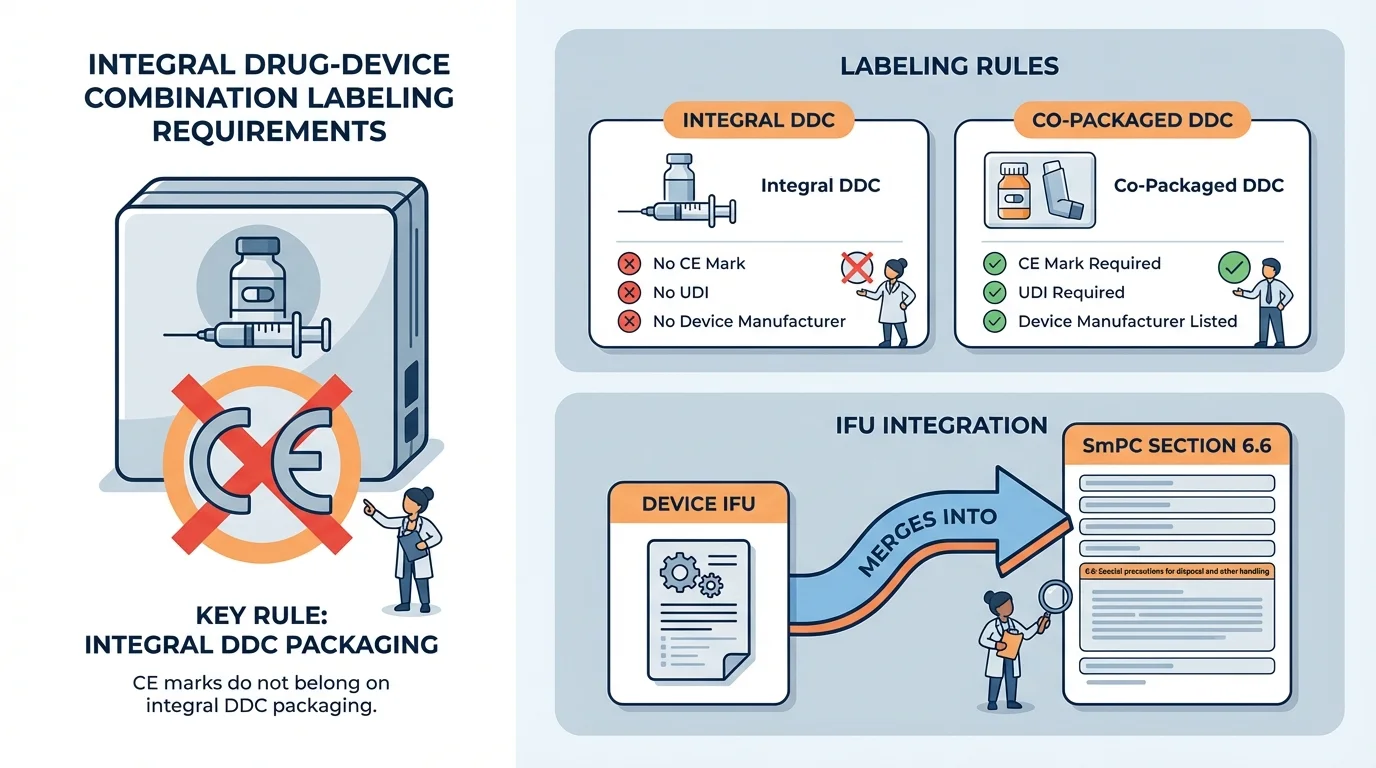
The EMA Q&A addresses labeling for integral DDCs within a broader section on labeling requirements. The core principle is straightforward: integral DDCs form a single integral product governed by pharmaceutical legislation-either Directive 2001/83/EC or Regulation (EC) No 726/2004. Because the product is regulated as a medicinal product, the labeling framework is pharmaceutical, not device.
This has several practical consequences that catch teams off guard, particularly those with device backgrounds. MDR obligations like CE marking are not required and should not appear on the iDDC packaging. The Quality Review of Documents (QRD) templates-not MDR Annex I-govern the labeling structure. And device administrative information (manufacturer name, notified body number, device symbols) stays off the medicinal product labeling entirely.
For integral DDCs, the product labelling should follow the requirements for medicinal products, as outlined in the QRD templates. MDR obligations such as the CE mark are not required and should not be applied to the package of such an iDDC.
Why Pharmaceutical Labeling Takes Precedence
Why this matters: The rationale behind this rule isn't arbitrary-it follows directly from how integral DDCs are classified. Understanding the logic prevents the instinct to "add device info just to be safe."
The reasoning follows a chain that starts with classification under Q1. An integral DDC is a single product where the drug and device form an inseparable unit. When the principal mode of action is pharmacological (the majority of integral DDCs), the product is regulated as a medicinal product under pharmaceutical legislation.
Labeling follows regulation. If the product is regulated as a medicinal product, its labeling must conform to medicinal product rules. Applying MDR labeling elements-CE marks, device manufacturer details, MDR symbols-would misrepresent the product's regulatory status. It would suggest to healthcare professionals and patients that the product has undergone conformity assessment as a medical device, when in reality the product as a whole has been assessed and authorized as a medicinal product.
This doesn't mean the device component is unregulated. The device part must still meet relevant General Safety and Performance Requirements (GSPRs) from MDR Annex I. It's assessed through the Article 117 consultation, where a Notified Body evaluates the device component's conformity. But that assessment happens within the pharmaceutical authorization process-and the labeling reflects the pharmaceutical authorization, not the device conformity assessment.
Labeling Framework: Integral DDC vs Co-Packaged DDC
| Element | Integral DDC (iDDC) | Co-Packaged DDC |
|---|---|---|
| Governing Framework | Pharmaceutical legislation (Directive 2001/83/EC) | Both pharmaceutical + MDR (separate for each component) |
| CE Mark on Package | No - must not appear on iDDC packaging | Yes - required on device or device packaging |
| UDI on Outer Packaging | No - should not appear on medicinal product labeling | Yes - required on device per MDR |
| Labeling Template | QRD template (pharmaceutical) | QRD for drug + MDR Annex I for device |
| Device Manufacturer on Label | No - only MAH details appear | Yes - device manufacturer must be identified |
| IFU Handling | Integrated into SmPC and Package Leaflet | Separate device IFU (may be combined for small devices) |
The CE Mark and UDI Rules for Integral DDCs
Why this matters: This is where the most common labeling errors occur. Teams with device experience instinctively add CE marks and UDI carriers-both of which should be absent from iDDC packaging.
The Q&A is explicit on this point: CE marks should not be applied to the package of an integral DDC. The reasoning is simple-CE marking signifies that a product has undergone conformity assessment as a medical device under MDR. An integral DDC has not. It has undergone marketing authorization as a medicinal product, and its regulatory status is represented by the marketing authorization number, not a CE mark.
CE Mark Decision for Integral DDC Components
Question
Does the device part of your iDDC already carry a CE mark?
No CE mark anywhere - straightforward
The device was developed specifically for this integral combination. No CE marking applies to the device or the product.
CE mark may remain on device - but must not appear on iDDC labeling or packaging
If the device was previously CE marked (e.g., a syringe used in other contexts), the mark on the device itself does not need to be removed. But it must not appear on the outer packaging, labeling, or product information of the iDDC.
UDI may remain on device - but must not appear on iDDC labeling
Similar to CE marks: if UDI is physically marked on the device component, it doesn't need removal. But UDI should not appear on the labeling, outer package, or product information of the integral combination product.
No CE mark anywhere - straightforward
The device was developed specifically for this integral combination. No CE marking applies to the device or the product.
CE mark may remain on device - but must not appear on iDDC labeling or packaging
If the device was previously CE marked (e.g., a syringe used in other contexts), the mark on the device itself does not need to be removed. But it must not appear on the outer packaging, labeling, or product information of the iDDC.
UDI may remain on device - but must not appear on iDDC labeling
Similar to CE marks: if UDI is physically marked on the device component, it doesn't need removal. But UDI should not appear on the labeling, outer package, or product information of the integral combination product.
The practical summary: What's physically on the device component can stay. What appears on the product's labeling, packaging, and product information must follow pharmaceutical rules exclusively. The distinction is between the device itself and the product as a whole-and for labeling purposes, only the product-level regulatory status matters.
Instructions for Use: Where Device Meets Drug
Why this matters: This is where the two regulatory worlds genuinely overlap. The device IFU can't be ignored-it must be translated into the pharmaceutical labeling framework. Getting this wrong creates medication error risks that assessors take very seriously.
The device component of an integral DDC has Instructions for Use-information about how to handle, prepare, and operate the delivery mechanism. For a pre-filled syringe, that's injection technique. For an autoinjector, that's activation sequence. For a transdermal patch, that's application and removal. This information is essential for patient safety, and the EMA's position is clear: it must be incorporated into the medicinal product's SmPC and Package Leaflet.
The key phrase from the Q&A is "clear and simple instructions for the intended use of the DDC, aimed at patients and/or healthcare professionals, written in such a way as to prevent medication errors." This isn't a suggestion-it's a requirement that reviewers actively check during assessment.
Integrating Device IFU into Pharmaceutical Labeling
- 1
Obtain Complete Device IFU
Get the full Instructions for Use from the device manufacturer. This is your source document for integration-every safety-relevant instruction must transfer to the pharmaceutical labeling.
- 2
Map IFU Content to QRD Sections
Identify which QRD template sections accommodate device instructions: Section 3 (How to use) in the Package Leaflet is the primary location. SmPC Section 6.6 (Special precautions for disposal and handling) covers preparation instructions.
- 3
Translate Device Language to Patient Language
Device IFUs are often written in technical language. Pharmaceutical Package Leaflets are written for patients. The content must transfer-but the language must be simplified and aligned with QRD readability standards.
- 4
Ensure Consistency Between IFU and PL
The instructions in the Package Leaflet must be consistent with the original device IFU. Any discrepancies-different step sequences, missing safety warnings, contradictory instructions-will be flagged during review.
- 5
Establish IFU Change Management
Any future changes to the device IFU must be reflected in the SmPC and Package Leaflet through pharmaceutical variation procedures. Contractual agreements between MAH and device manufacturer should govern how IFU changes are communicated.
The Integral DDC Labeling Checklist
Why this matters: Labeling deficiencies are among the most common reasons for Day 120 questions in centralized procedures. This checklist covers the elements assessors verify.
Integral DDC Labeling Verification
- Product labeling follows QRD template (not MDR Annex I labeling)
- No CE mark on outer packaging or product labeling
- No UDI on outer packaging or product information
- No device manufacturer administrative details on product labeling
- No device-specific symbols (MDR symbols) on product labeling
- Device IFU content integrated into Package Leaflet Section 3
- Device handling instructions included in SmPC Section 6.6
- Instructions written for target user (patient-friendly language)
- Consistency verified between device IFU and Package Leaflet instructions
- Change management process established for future IFU updates
- Full device instructions incorporated with finished product
Labeling Traps That Delay Submissions
Why this matters: These aren't theoretical risks-they're the patterns that generate Day 120 questions, List of Questions, and labeling-related review cycles in real submissions.
Adding CE Mark "Just in Case"
Teams with device backgrounds sometimes include the CE mark on iDDC packaging as a precaution-reasoning that it can't hurt. It can. The CE mark on an iDDC package misrepresents the product's regulatory status and will generate an explicit deficiency from the assessor.
Copying the Device IFU Verbatim into the Package Leaflet
Device IFUs and pharmaceutical Package Leaflets serve different audiences with different reading levels. Pasting the device IFU into the PL without adapting the language creates readability issues and fails QRD compliance. The content must transfer; the language must translate.
No Change Management for Device IFU Updates
The device manufacturer updates the IFU. The update doesn't reach the MAH. The Package Leaflet now contains outdated device instructions. This creates a safety signal that assessors take seriously. Contractual agreements must ensure IFU changes trigger pharmaceutical variation procedures.
Including Device Manufacturer Details on Product Labeling
For iDDCs, only the MAH's details appear on the product labeling. Including the device manufacturer's name, address, or notified body number on the product information violates the labeling framework and implies a separate device assessment pathway that doesn't apply.
FAQ
Should the CE mark appear on the packaging of an integral drug-device combination?
Do UDI requirements apply to integral drug-device combinations?
How should the Instructions for Use for the device part be handled in an integral DDC?
Which labeling template governs integral DDC labeling?
What happens if the device component of an integral DDC changes its IFU after marketing authorization?
Would Your iDDC Labeling Pass Review?
One misplaced CE mark. One missing IFU section. That's all it takes to trigger a labeling deficiency. Two minutes to check your approach.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Assess Your Labeling →5 questions · Personalized insights · Free