EMA Regulatory
Article 117 Consultation Procedure: EMA Opinion for Drug-Device Combinations
Quick Answer
Article 117 consultation is the procedure by which the EMA/CHMP provides an opinion on the quality and safety of an ancillary medicinal substance incorporated into a medical device. Required since 26 May 2021, it follows a 210-day timeline and the Notified Body-not the manufacturer-is the applicant.
You're reviewing a submission timeline for a drug-eluting stent. Someone asks: "Do we need EMA consultation before the Notified Body can issue the CE certificate?"
You pause. The device incorporates a medicinal substance where the action is ancillary to the device-but you're not sure whether that means you need to consult EMA first, or whether the Notified Body handles it independently.
Get this wrong, and your timeline extends by months. Get it right, and you can plan your pre-submission activities with confidence.
What Is the Article 117 Consultation?
Article 117 of the Medical Devices Regulation (EU) 2017/745 introduced amendments to Annex I, Section 3.2(12) of Directive 2001/83/EC. These amendments created a mandatory consultation procedure for certain drug-device combinations.
The core purpose: To ensure that when a medical device incorporates a medicinal substance with ancillary action, the EMA or a national competent authority provides a scientific opinion on the quality and safety of that substance-including its clinical benefit/risk profile.
Key distinction: This applies to medical devices with incorporated medicinal substances-where the device is the primary product and the drug action is ancillary. This is the opposite of integral DDCs regulated under pharmaceutical legislation, where the medicinal product is primary.
Products That Require Article 117 Consultation
Article 117 consultation applies to devices described in MDR Articles 1(8) and 1(9), specifically:
Article 1(8) Devices
Devices that incorporate a substance which, if used separately, would be considered a medicinal product-and which has ancillary action to the device.
Examples: Drug-eluting stents, bone cements with antibiotics, heparin-coated catheters
Article 1(9) Devices
Devices that incorporate a human blood derivative, which has ancillary action to the device.
Examples: Surgical sealants containing fibrin, haemostatic devices with human plasma components
When Is Article 117 Consultation Required?
Why this matters: Not every device with an incorporated substance requires EMA consultation. Understanding the triggers prevents unnecessary procedures-and catches requirements you might miss.
Article 117 consultation is required when a Notified Body is conducting conformity assessment for a device that incorporates an ancillary medicinal substance or human blood derivative, and the Notified Body needs an opinion on the quality and safety of that substance.
The Effective Date
Article 117 requirements apply to conformity assessment procedures initiated on or after 26 May 2021-the date the MDR came into full application.
Question
Does Your Device Require Article 117 Consultation?
Yes - EMA or national CA consultation required
Yes - EMA consultation required
No - Regulated as medicinal product instead
No - Separate Article 9 pathway applies
The key trigger is whether the incorporated substance's action is ancillary to the device. If the substance's action is principal, the product is regulated as a medicinal product-not a device-and follows pharmaceutical legislation instead.
What "Ancillary" Means
A substance has ancillary action when it supports, enhances, or complements the device's primary mode of action-but the device could still function (perhaps less effectively) without it.
| Substance Role | Example | Regulatory Pathway |
|---|---|---|
| Ancillary action | Antibiotic coating on orthopaedic implant | MDR + Article 117 consultation |
| Principal action | Drug in a pre-filled syringe delivery system | Pharmaceutical legislation (Directive 2001/83/EC) |
Post-Authorisation Changes
Article 117 consultation doesn't just apply at initial conformity assessment. It also applies post-certification if there's a major change to the incorporated substance that could impact:
- The source of the substance
- The manufacturing process
- The amount or concentration
- The method of incorporation
- Any factor affecting quality, safety, or efficacy
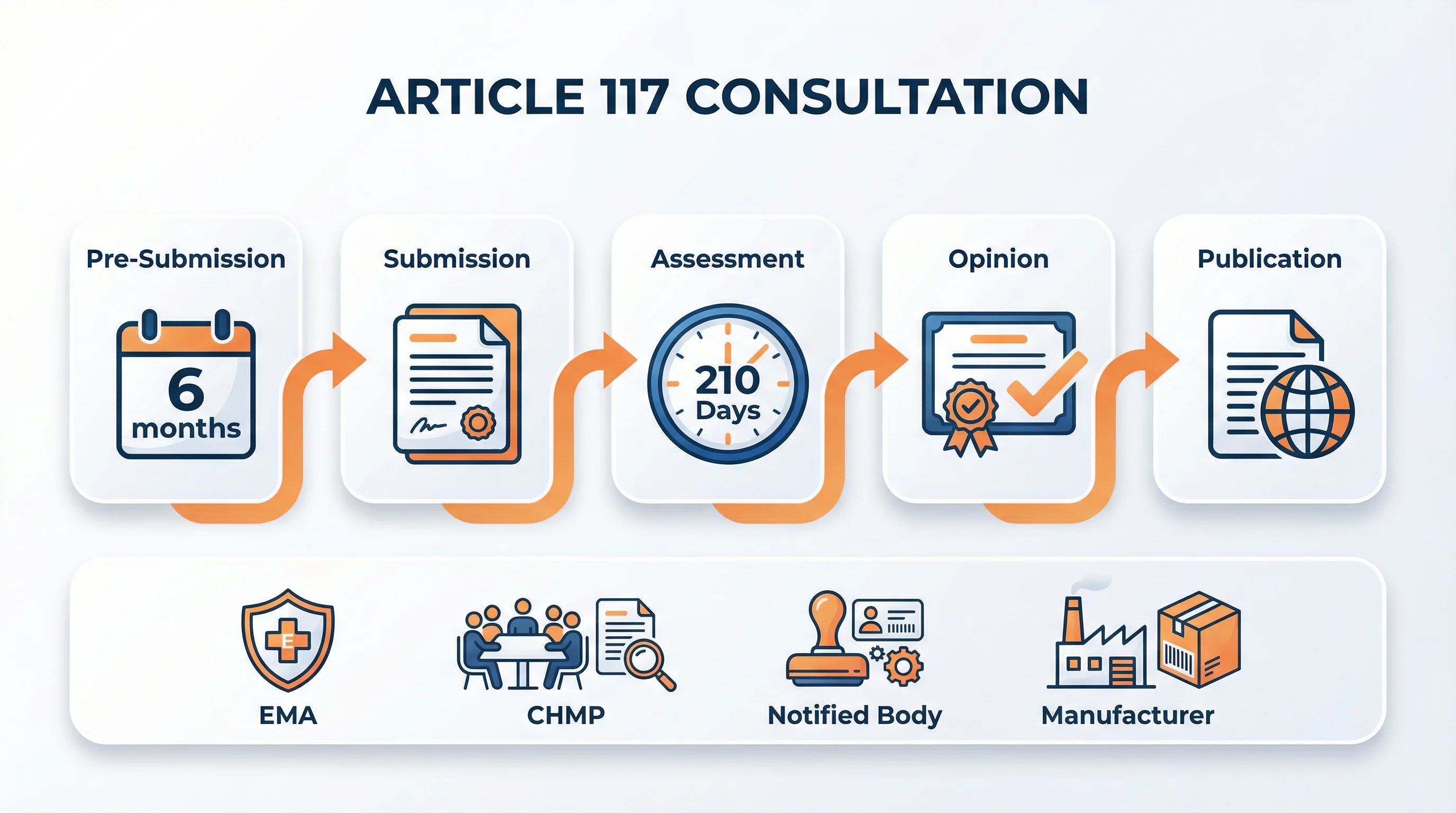
Procedure and Timeline: 210 Days Explained
Why this matters: The 210-day timeline doesn't run continuously. Understanding clock stops and pre-submission planning is essential for accurate project timelines.
Article 117 Consultation Procedure
- 1
Pre-Submission
Notified Body provides intention letter 6+ months before submission. Device manufacturer collaborates on dossier preparation.
- 2
Submission
Notified Body submits consultation application to EMA using official application form.
- 3
Validation
EMA validates completeness. CHMP appoints one or two rapporteurs.
- 4
Assessment
210-day evaluation with clock stops for responses to questions or deficiencies.
- 5
Opinion
CHMP/EMA issues opinion on quality, safety, and benefit/risk profile.
- 6
Publication
Public assessment report (CPAR) published within 3 months of opinion.
The 210-day timeline mirrors the Centralised Procedure for medicinal products. Clock stops occur when the applicant (Notified Body) needs time to respond to questions-these periods don't count against the 210 days.
Pre-Submission Activities
EMA strongly encourages pre-submission engagement. The key elements:
Intention to Submit Letter
The Notified Body should provide this letter at least 6 months before the expected submission date. It must include:
- → Expected submission date
- → Scientific explanation demonstrating that the medicinal substance's action is ancillary to the device
- → Overview of the device and incorporated substance
Accelerated Assessment
In specific circumstances, accelerated assessment may be requested. Qualifying conditions include:
- Devices used in serious diseases (life-threatening or heavily disabling)
- Known medicinal substance or human blood derivative from a known source, where the CHMP deems the evaluation less extensive
Justification for accelerated assessment should be provided at least 10 working days before the CHMP meeting preceding the intended start of the procedure.
Documentation Requirements
Why this matters: Incomplete documentation triggers clock stops and delays. Knowing exactly what's required upfront prevents avoidable timeline extensions.
Article 117 Consultation Dossier Requirements
- Scientific explanation that substance action is ancillary to device
- Detailed description of the device and intended purpose
- Characterisation of the ancillary medicinal substance or human blood derivative
- Manufacturing information for the substance - including source and quality controls
- Non-clinical data supporting safety of the substance
- Clinical data on the benefit/risk profile of incorporation
- Documentation of substance-device interaction
- Proposed labelling and instructions for use
The dossier is prepared collaboratively between the device manufacturer and the Notified Body. The Notified Body takes responsibility for submission, but the manufacturer provides the underlying technical documentation.
Quality and Safety Data
The EMA assessment focuses on whether the incorporated substance meets quality and safety standards. Key areas of scrutiny include:
Quality Assessment
- • Source and origin documentation
- • Manufacturing process validation
- • Specifications and acceptance criteria
- • Stability data for the incorporated form
- • Impurity characterisation
Safety Assessment
- • Toxicological profile
- • Local and systemic effects
- • Pharmacological interactions
- • Biocompatibility considerations
- • Clinical benefit/risk analysis
What Cannot Be Accepted
EMA guidance is clear on several documentation requirements:
- Partial compliance: A Notified Body opinion or compliance statement concluding on partial compliance with GSPRs cannot be accepted-full compliance is required
- Cross-references to national dossiers: It's not possible to cross-refer to the dossier of nationally authorised medicinal products
- Scope misalignment: The scope of the Notified Body opinion must align with the intended purpose of the device for the specific consultation
Roles: EMA, CHMP, and Notified Bodies
Why this matters: Understanding who does what prevents miscommunication and ensures you're coordinating with the right parties at each stage.
European Medicines Agency (EMA)
- • Receives and validates consultation applications
- • Manages the procedural aspects and timeline
- • Issues the scientific opinion on quality and safety
- • Publishes the Consultation Public Assessment Report (CPAR)
- • Provides advice to Notified Bodies on post-certification changes
- • Manages fees for the consultation procedure
Committee for Medicinal Products for Human Use (CHMP)
- • Appoints one or two rapporteurs to lead the evaluation
- • Follows the 210-day assessment timetable
- • Can grant accelerated assessment in qualifying cases
- • Issues the scientific opinion on the ancillary substance
- • For ATMPs, the Committee for Advanced Therapies (CAT) handles this role
Notified Bodies
- • Acts as the applicant for the consultation procedure
- • Prepares the consultation dossier with the device manufacturer
- • Provides the intention to submit letter
- • Responds to questions during the assessment period
- • Takes the EMA/CHMP opinion into account when making their decision on CE marking
- • Informs EMA and consults on post-certification changes
Post-Consultation: Managing Changes
Article 117 doesn't end with the initial opinion. Changes to the incorporated substance after CE marking require ongoing communication with EMA.
When to Consult EMA on Changes
If changes are made to an ancillary medicinal substance or blood derivative incorporated in the device, the Notified Body must:
- Inform EMA of the proposed change
- Consult EMA to confirm that the quality and safety of the ancillary substance are maintained
- Receive updated scientific opinion if EMA determines the change could impact the benefit/risk profile
The Notified Body must consider any updated EMA opinion in its ongoing conformity assessment. This creates a continuous regulatory relationship, not a one-time consultation.
Changes That Trigger Consultation
| Change Type | EMA Consultation Required? |
|---|---|
| Change in source or origin of substance | Yes |
| Change in manufacturing process | Yes |
| Change in amount or concentration | Yes |
| Change in method of incorporation | Yes |
| Administrative changes only | Case-by-case |
Practical Guidance for Regulatory Affairs
For Device Manufacturers
- 1. Engage early with your Notified Body - They're the applicant, so alignment is essential
- 2. Prepare comprehensive documentation - The dossier is collaborative, but you provide the technical substance
- 3. Plan for 210+ days - Include clock stops in your timeline; 18 months total is realistic
- 4. Establish change management processes - Any modification to the substance triggers EMA notification
- 5. Request pre-submission meeting - EMA encourages this at least 6 months before submission
For Notified Bodies
- • Provide intention to submit letter 6+ months before expected submission date
- • Include scientific justification that substance action is ancillary
- • Collaborate closely with device manufacturer on dossier preparation
- • Take EMA/CHMP opinion into account for CE marking decision
- • Maintain ongoing communication with EMA for post-certification changes
Timeline Planning Tips
- • Minimum realistic timeline: 18 months from intention letter to CE decision
- • Pre-submission activities: 6 months minimum
- • Assessment period: 210 days (plus clock stops)
- • NB decision post-opinion: Variable, but typically 2-3 months
- • Buffer for questions: Add 3-6 months for clock stops
Key Regulatory Milestones
26 May 2021
MDR came into full application; Article 117 consultation requirements apply
210 Days
Maximum CHMP assessment period (excluding clock stops)
6 Months
Recommended lead time for intention to submit letter
References
- 1. Regulation (EU) 2017/745 on medical devices (MDR), Article 117
- 2. EMA Recommendation on Procedural Aspects and Dossier Requirements for Consultation
- 3. EMA Q&A on implementation of medical devices and IVD regulations
- 4. Directive 2001/83/EC, Annex I, Section 3.2(12) as amended
- 5. EMA Procedural Advice on Consultation for Combined ATMPs
Need faster answers on EMA procedures? RegulatorySense delivers instant, authoritative guidance with citations.
FAQ
What is the purpose of Article 117 consultation?
When did Article 117 consultation requirements come into effect?
How long does the Article 117 consultation take?
Who is the applicant for Article 117 consultation?
Does Article 117 apply to combined ATMPs?
Uncertain About Your Article 117 Documentation?
Article 117 consultations require precise coordination between Notified Bodies, manufacturers, and EMA. Take our 2-minute assessment to discover where gaps might exist in your regulatory research confidence.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Take the Assessment →5 questions · Personalized insights · Free