EMA Regulatory
DDC Approval Timeline: Every Milestone from Start to Finish
Quick Answer
EU drug-device combination approval typically takes 3-5 years end-to-end. The critical milestones are Notified Body engagement (12-18 months before MAA), NB Opinion issuance, MAA submission, 210-day EMA assessment with two mandatory clock stops around Day 120 and Day 180, CHMP opinion, and EU Commission decision within 67 days. Clock stops alone typically add 6-12 months beyond the 210-day assessment period.
Her competitor's drug-autoinjector combination had just received EU marketing authorization. The press release went out on a Tuesday morning, and by that afternoon, three people had forwarded it to her with the same question: how far behind are we?
She opened her project plan and started counting backward from the competitor's first public mention of Notified Body engagement. Twenty-eight months. That's what separated the start of their regulatory journey from the authorization announcement.

Twenty-eight months-and that was a relatively smooth pathway. No extended clock stops. No Notified Body capacity delays. No major objections during the CHMP assessment. For a product with complications at any of those stages, the timeline stretches well beyond three years.
What follows is the complete milestone map for EU drug-device combination approval. Not theoretical ranges, but the actual sequence of events, with realistic durations at each stage and the points where timelines most often slip.
This is the twelfth and final guide in our Fundamentals series. If you've followed the series from the beginning, you now have the complete foundation for understanding EU drug-device combination regulation-from classification to approval timeline.
The Complete DDC Approval Timeline
Why this matters: Seeing the entire timeline in one view reveals the dependencies between stages-and shows why delays at early milestones cascade through the entire approval process.
Drug-Device Combination Approval: End-to-End Timeline
Month 0-3
Notified Body Selection & Engagement
Identify MDR-designated NB with relevant scope. Submit expression of interest, negotiate contract. Limited NB capacity means this step alone can take 3-6 months.
Month 3-9
Device Documentation Preparation
Compile GSPR compliance documentation, risk management files, design documentation, biocompatibility data, and usability engineering reports for the device component.
Month 9-15
Notified Body Assessment & Opinion
NB reviews device documentation, may request clarifications. Issues NB Opinion report on device conformity with MDR Annex I GSPRs.
Month 15-18
MAA Dossier Finalization
Integrate NB Opinion into Module 3.2.R quality documentation. Complete pharmaceutical, preclinical, and clinical dossier sections.
Month 18
MAA Submission
Submit complete marketing authorization application to EMA (centralized) or national authority. Day 0 of the 210-day assessment clock.
Month 18-22 (Day 1-120)
First Assessment Phase
Rapporteur and Co-rapporteur assess the dossier. Day 80: preliminary assessment reports. Day 120: CHMP issues list of questions (LoQ). CLOCK STOP 1 begins.
Month 22-28 (Clock Stop 1)
Applicant Responds to Day 120 Questions
No fixed duration-typically 3-6 months. Applicant prepares written responses, potentially supplemented by oral explanation. Clock resumes upon response submission.
Month 28-31 (Day 121-180)
Second Assessment Phase
CHMP evaluates responses. Day 150: Joint assessment report. Day 180: Outstanding issues identified. CLOCK STOP 2 begins if needed.
Month 31-34 (Clock Stop 2)
Final Clarifications
Typically 1-3 months. Addresses remaining concerns. Oral explanation may be requested.
Month 34-36 (Day 181-210)
CHMP Opinion
Final CHMP discussion and vote. Positive opinion triggers Commission decision process. Negative opinion can be re-examined within 15 days.
Month 36-38
EU Commission Decision
Commission grants marketing authorization within 67 days of CHMP opinion. Marketing authorization valid in all EU/EEA member states.
Month 38-42
National Implementation & Launch
Pricing and reimbursement negotiations (country-specific). Supply chain preparation. First patient access.
3-5 years
typical end-to-end timeline
From Notified Body engagement to first patient access for drug-device combinations in the EU
Pre-Submission Phase (12-24 Months Before MAA)
Why this matters: The pre-submission phase is where timelines are won or lost. Starting NB engagement too late is the single most common cause of DDC approval delays.
Pre-Submission Critical Path
- 1
NB Selection
Check NANDO database for MDR-designated NBs with scope for your device type. Assess capacity, proposed timelines, and geographic considerations.
- 2
Contract & Scope
Define assessment scope, deliverables, and timeline expectations. Negotiate data-sharing arrangements with device suppliers if applicable.
- 3
Documentation Assembly
Prepare device technical documentation per MDR Annex II. Include GSPR checklist, risk management file, usability engineering, and clinical data.
- 4
NB Submission
Submit documentation package to NB. Respond to clarification requests promptly-each delay extends the NB Opinion timeline.
The reduced number of MDR-designated Notified Bodies-compared to the MDD era-means capacity constraints are real. Some NBs have waiting lists of 6 months or more before they can begin an assessment. Factor this into your planning: the day you decide to pursue EU approval is not the day your NB timeline begins.
Pro Tip
Establish contact with potential Notified Bodies during clinical development, not after. Even preliminary discussions about scope and capacity can prevent months of delay when you're ready to submit device documentation.
Teams that engage NBs during Phase 3 clinical trials consistently achieve earlier MAA submissions than those who start after Phase 3 readout.
Obtaining the Notified Body Opinion
Why this matters: The NB Opinion is a prerequisite for your MAA dossier. Its timing directly controls your MAA submission date-and therefore your entire downstream timeline.
The Notified Body conducts an independent assessment of the device component against MDR Annex I General Safety and Performance Requirements. This is not a rubber-stamp exercise-NBs perform substantive technical review and may require multiple clarification rounds before issuing their opinion.
Milestone Readiness: NB Opinion Submission
- GSPR compliance checklist complete with supporting evidence for each requirement
- Risk management file per ISO 14971, including benefit-risk analysis
- Usability engineering file per IEC 62366-1 with summative evaluation results
- Biocompatibility evaluation per ISO 10993 series for patient-contacting components
- Design verification and validation documentation complete
- Supplier data-sharing agreements in place for third-party device components
- Labeling and IFU drafts aligned with both MDR Annex I Chapter III and pharmaceutical requirements
Common Timeline Slip: Supplier Data Gaps
When the device component involves third-party manufactured parts (e.g., a pen injector mechanism from a device OEM), obtaining the technical documentation needed for the NB assessment can take months. Contractual data-sharing arrangements should be established during commercial negotiations, not during regulatory preparation.
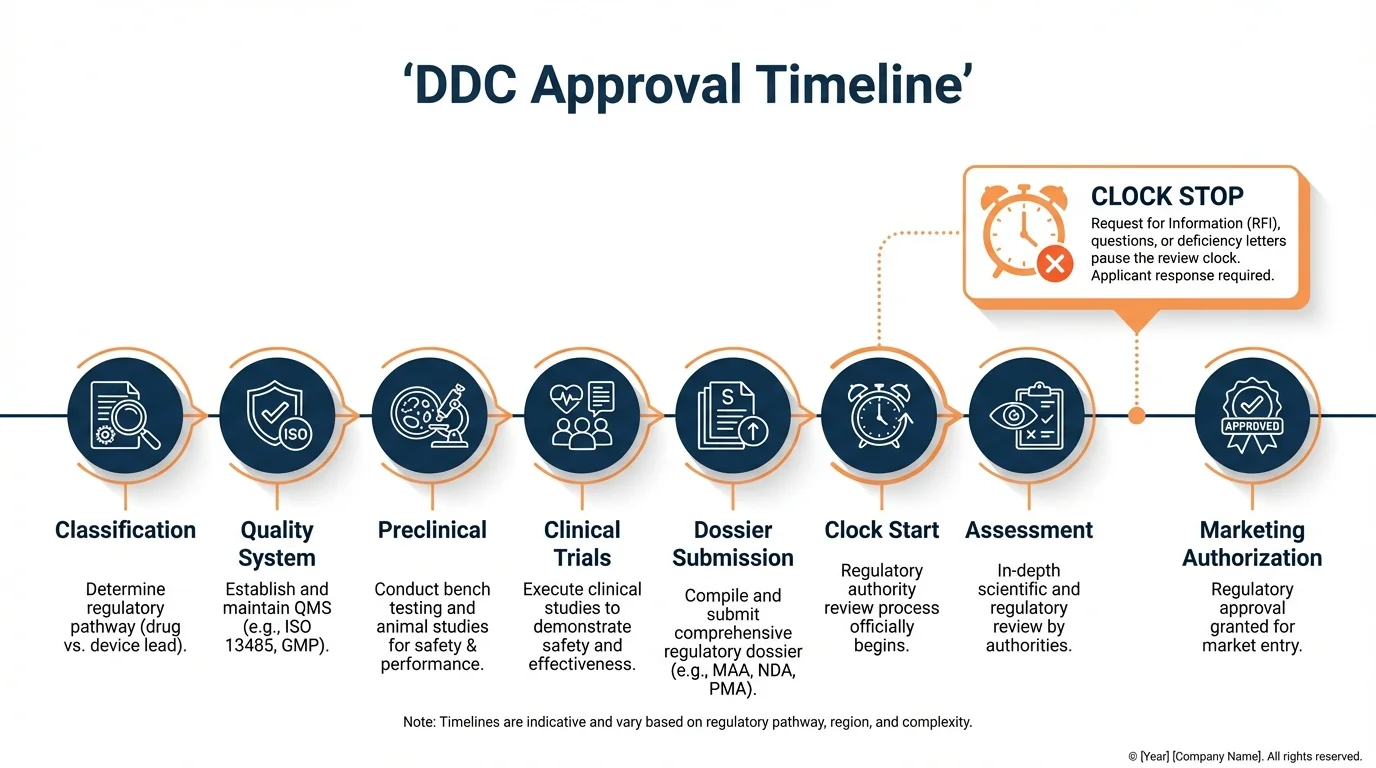
MAA Assessment: 210 Days Plus Clock Stops
Why this matters: The 210-day assessment period is what most teams plan around, but it's the clock stops that actually determine total assessment duration. Planning only for 210 days will leave your launch timeline months short of reality.
The centralized procedure allocates 210 active assessment days, divided into distinct phases. A Rapporteur and Co-rapporteur from different EU member states lead the evaluation, producing assessment reports at key intervals that the full CHMP reviews.
Phase 1: Days 1-120
- Day 1: Assessment clock starts on MAA validation
- Day 80: Preliminary assessment reports from Rapporteur and Co-rapporteur
- Day 120: CHMP adopts List of Questions (LoQ) and List of Outstanding Issues (LoOI)
- Clock Stop 1 begins: Applicant receives questions for response
Phase 2: Days 121-180
- Day 121: Clock resumes when applicant submits responses to Day 120 questions
- Day 150: Joint Rapporteur/Co-rapporteur assessment of responses
- Day 180: CHMP identifies any outstanding issues remaining
- Clock Stop 2 (if needed): Final clarifications requested
Phase 3: Days 181-210
- Day 181: Clock resumes for final phase
- Day 210: CHMP adopts opinion (positive, negative, or request for re-examination)
- Oral explanations may be requested at any point during Phase 2 or 3
Understanding Clock Stops
Why this matters: Clock stops are the most unpredictable element of the DDC approval timeline. A single extended clock stop can add 6 months to your approval date, and they're entirely within the applicant's control to minimize.
During a clock stop, the 210-day assessment timer pauses while the applicant prepares responses. There is no regulatory maximum for clock stop duration-it ends when the applicant submits adequate responses. In practice, clock stops for complex DDC submissions tend to be longer than for straightforward pharmaceutical products because questions often span both drug and device domains.
Clock Stop 1 (Day 120)
- • Typically 3-6 months duration
- • Major scientific questions addressed
- • May include device-specific GSPR questions
- • Additional clinical data may be requested
- • GMP inspection scheduling often begins
Clock Stop 2 (Day 180)
- • Typically 1-3 months duration
- • Outstanding issues from Phase 2
- • Final labeling and product information
- • Risk management updates if required
- • Oral explanation may be scheduled
Pro Tip
Prepare a 'response war room' team before Clock Stop 1 begins. Having device regulatory, pharmaceutical regulatory, clinical, quality, and CMC experts available immediately when questions arrive can cut response preparation time by weeks.
The clock stop duration is largely applicant-driven. Fast, comprehensive responses directly shorten your total approval timeline.
Post-Opinion to Market Access
Why this matters: A positive CHMP opinion isn't market access. Several more milestones remain before patients can receive your product-and national pricing and reimbursement negotiations can add another 12-24 months in some EU markets.
After a positive CHMP opinion, the EU Commission has 67 days to grant the marketing authorization. This is largely procedural-the Commission follows the CHMP recommendation in the vast majority of cases. The marketing authorization is then valid across all EU/EEA member states simultaneously.
However, market access requires additional national steps that vary by member state. Pricing and reimbursement negotiations, supply chain establishment, and healthcare professional education all add to the interval between authorization and first patient access.
Strategies to Protect Your Timeline
Why this matters: You can't eliminate regulatory timelines, but you can protect against the most common causes of delay. These strategies are the difference between a 30-month pathway and a 48-month pathway.
Early NB Engagement (Saves 3-6 Months)
Engage your Notified Body during Phase 2/3 clinical development, not after data readout. Early scope discussions, preliminary document reviews, and capacity reservation can prevent the most common DDC timeline delay. The NB Opinion is on the critical path-every month of NB delay pushes your MAA submission by the same amount.
Pre-Submission Meetings with EMA (Saves 2-4 Months)
Scientific advice from EMA on DDC-specific issues-including device documentation expectations, clinical evidence requirements, and Article 117 documentation-reduces the likelihood of major objections during assessment. Fewer major questions means shorter clock stops.
Parallel Documentation (Saves 3-6 Months)
Prepare the device documentation package in parallel with clinical development, not sequentially after. Design verification and validation, usability studies, and biocompatibility testing can proceed alongside Phase 3 trials.
Clock Stop Response Readiness (Saves 1-3 Months)
Anticipate likely Day 120 questions based on similar DDC assessments and EMA's published assessment reports. Pre-draft responses to predictable questions so your team can finalize rapidly when the actual List of Questions arrives.
References
- 1. EMA Procedural Timetables for Centralized Procedure (standard and accelerated)
- 2. Regulation (EU) 2017/745 (MDR), Article 117 and Annex I (GSPRs)
- 3. EMA Questions and Answers on implementation of MDR and IVDR (Revision 5, January 2025)
- 4. EC Regulation 726/2004 - Centralized Authorization Procedure
- 5. EMA Guidance on Marketing Authorization Application - Module 3.2.R Quality Documentation for DDCs
- 6. Directive 2001/83/EC as amended by MDR Article 117 - Marketing Authorization Requirements for DDCs
Need faster answers? RegulatorySense delivers instant, authoritative guidance with source citations.
FAQ
How long does it take to approve a drug-device combination product in the EU?
What is a clock stop in the EU marketing authorization process?
When should I engage a Notified Body for a drug-device combination product?
Can I get accelerated assessment for a drug-device combination product?
Would Your Timeline Survive a Clock Stop?
Clock stops add months. Notified Body delays add more. Two minutes to stress-test whether your plan accounts for the milestones that actually drive DDC timelines.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Stress-Test Your Plan →5 questions · Personalized insights · Free