EMA Regulatory
Drug-Device Combination Products Under EU MDR: The Complete Regulatory Guide
Quick Answer
Drug-device combinations under EU MDR are medicinal products that incorporate devices for administration, delivery, or dosing. The regulatory pathway depends on whether it's integral (single product) or non-integral (separate packaging). Integral DDCs with Class IIa+ devices require Notified Body opinion.
The board presentation is in two hours. The VP leans in and asks: "Does our new autoinjector need a separate CE mark, or can we handle it through the MAA?"
You pause. You think you know the answer. But MDR Article 117 is a maze, and the consequences of getting this wrong are a Complete Response Letter and six months of delay.
The path forward: a framework that traces each requirement back to its legal source.
What Is a Drug-Device Combination Product?
A drug-device combination product (DDC) is a medicinal product that incorporates one or more medical devices as part of its composition or administration system.
The EMA defines DDCs as:
"Medicinal products which contain one or more medical device(s) as an integral part of the composition, as well as medicinal products for which one or more medical device(s) and/or device component(s) are necessary for use of the medicinal product."
Common Examples of Drug-Device Combinations
Integral DDCs
(device and drug physically combined)
- • Pre-filled syringes and pens
- • Autoinjectors
- • Dry powder inhalers
- • Transdermal patches
- • Drug-releasing implants
- • Nasal and oromucosal sprays
Non-Integral DDCs
(device co-packaged or referenced)
- • Oral administration devices (cups, spoons, syringes)
- • Injection needles
- • Refillable pens using cartridges
- • Nebulizers
- • Infusion pumps
The Regulatory Framework: MDR vs Pharmaceutical Legislation
The key question for any DDC is: Which regulatory framework applies?
The answer depends on the product's principal mode of action (PMOA).
If the Principal Mode of Action Is Pharmacological
The product is regulated as a medicinal product under Directive 2001/83/EC or Regulation (EC) No 726/2004.
However, the device component must still comply with the relevant General Safety and Performance Requirements (GSPRs) from MDR Annex I.
If the Principal Mode of Action Is Device-Based
The product is regulated as a medical device under Regulation (EU) 2017/745 (MDR).
If the device incorporates a medicinal substance with ancillary action, a scientific opinion from EMA or a national competent authority is required before the notified body can issue a CE certificate.
Article 117: The Critical Amendment for DDC Manufacturers
Article 117 of the MDR introduced significant changes to the marketing authorization requirements for integral DDCs.
What Article 117 Requires
For marketing authorization applications (MAAs) submitted on or after 26 May 2021, the dossier must include evidence of the device part's conformity with relevant GSPRs.
Three pathways to demonstrate conformity:
| Pathway | When to Use | Documentation Required |
|---|---|---|
| EU Declaration of Conformity | Device manufacturer has already assessed conformity | Manufacturer's declaration |
| EU Certificate from Notified Body | Higher-risk devices (Class Im, Is, IIa, IIb, III) | NB certificate allowing CE marking |
| Notified Body Opinion (NBOp) | No existing conformity assessment available | NB opinion on GSPR compliance |
When Is a Notified Body Opinion Required?
A Notified Body Opinion is required when:
- The dossier does not include results of a conformity assessment, AND
- If the device were used separately, it would require involvement of a notified body
Class I devices (excluding Im, Is, Irsi)
The applicant can self-declare conformity with GSPRs.
Class Im, Is, Irsi, IIa, IIb, and III devices
A Notified Body Opinion is mandatory if no EU certificate exists.
Two Types of Integral DDCs Under MDR
Why This Matters: This distinction determines your ENTIRE regulatory pathway. Get it wrong, and you're either over-documenting (wasting months) or risking rejection.
The MDR defines two types of integral drug-device combinations:
Type 1: Article 1(8) Second Subparagraph
Devices that incorporate, as an integral part, a substance that would be considered a medicinal product if used separately, where the action of that substance is principal.
Example: An ingestible sensor incorporated into a medicinal product where the therapeutic effect comes primarily from the drug, not the sensor.
Type 2: Article 1(9) Second Subparagraph
Medical devices intended to administer a medicinal product where they form a single integral product meeting three conditions:
- Device and medicinal product form a single integral product when placed on market
- Intended exclusively for use in the given combination
- Not reusable
Examples: Pre-filled syringes, pre-filled pens, nebulizers pre-charged with specific medicinal product, transdermal patches.
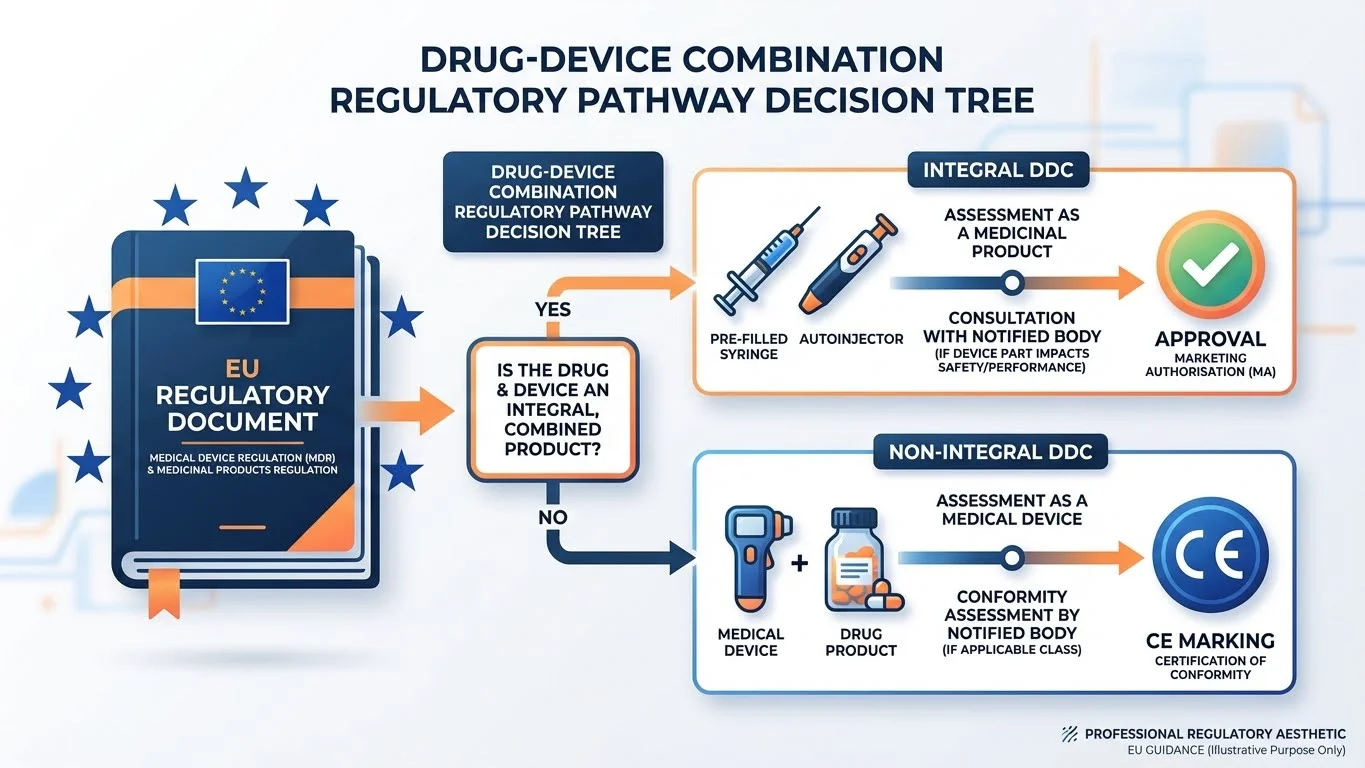
Integral vs Non-Integral DDCs: Key Differences
Whether your DDC is integral or non-integral determines the regulatory pathway and documentation requirements.
| Characteristic | Integral DDCs | Non-Integral DDCs |
|---|---|---|
| Physical integration | Combined during manufacturing | Separate but combined at point of use |
| CE marking | Not required on outer packaging | Required on the device |
| Conformity evidence | Via Article 117 pathway | EU DoC from device manufacturer |
| NBOp | Required for higher-risk classes | Not applicable |
Quality Documentation Requirements
Why This Matters: This is where most DDC submissions fail. Assessors look for specific documentation in Module 3.2.R-here's exactly what they expect.
The EMA's Guideline on quality requirements for drug-device combinations (EMA/CHMP/QWP/BWP/259165/2019) specifies what must be included in the marketing authorization dossier.
Module 3.2.P - Drug Product
- P.1: Description and composition of the DDC
- P.2: Pharmaceutical development including device selection rationale
- P.2.4: Container closure system development
- P.5: Control of drug product specifications
- P.7: Container closure system specifications
- P.8: Stability studies including device functionality
Module 3.2.R - Regional Information
This section must include:
- • EU Declaration of Conformity, OR
- • EU Certificate from Notified Body, OR
- • Notified Body Opinion
Plus:
- • Usability/human factors study summaries
- • Platform technology documentation (if applicable)
- • Cross-references to relevant 3.2.P sections
Usability and Human Factors Studies
Why This Matters: Human factors isn't optional for DDCs. It's the difference between an approval and a deficiency letter asking you to demonstrate safe use in your target population.
If the device:
- Has not been used in the proposed patient population before, OR
- Is being used in a new or different setting
A usability study is expected to demonstrate that the DDC can be used safely to deliver the medicinal product to the target population.
What Usability Studies Should Cover
- ✓ Dose delivery accuracy under realistic conditions
- ✓ User comprehension of instructions
- ✓ Error frequency and severity
- ✓ Patient population-specific considerations
Standards Reference: Applicants should follow IEC 62366-1:2015 (Application of usability engineering to medical devices) and IEC/TR 62366-2:2016 (Guidance on usability engineering).
Lifecycle Management Considerations
Changes to the device component after initial marketing authorization require careful consideration.
When Changes Require Variation Applications
Any change to the medical device or device component within a DDC must be submitted under the appropriate variation category based on:
- Impact on Critical Quality Attributes (CQAs)
- Impact on overall DDC control strategy
- Nature and magnitude of the change
Documentation Updates
Depending on the change, updates may be required to:
- • Notified Body Opinion
- • Declaration of Conformity
- • CE mark documentation
Risk Assessment Updates
Changes to the device may require:
- • Updated risk management documentation
- • Revised human factors/usability studies
- • Communication plans for patients and HCPs
Practical Steps for Regulatory Affairs Professionals
For New DDC Development
- 1. Determine PMOA - Is the principal mode of action pharmacological or device-based?
- 2. Classify the device - What MDR risk class applies?
- 3. Engage early - Seek scientific advice from EMA if the classification is unclear
- 4. Choose your pathway - EU certificate, Declaration of Conformity, or NBOp?
- 5. Select a notified body - Ensure they're designated for your device type (check NANDO database)
For Borderline Products
- • Consult national competent authorities (NCAs) for formal advice
- • Use EMA's Innovative Task Force (ITF) for informal scientific input
- • Review MDCG guidance documents
- • Test your knowledge with our regulatory confidence assessment
For Lifecycle Changes
- 1. Assess impact - Does the change affect device performance or CQAs?
- 2. Determine variation category - Consult CA if unclear
- 3. Update documentation - NBOp, conformity evidence, risk management
- 4. Plan communications - If instructions for use change, consider patient/HCP communication
Key Dates and Transitional Provisions
26 May 2021
MDR came into application; Article 117 requirements apply to new MAAs
26 May 2022
IVDR came into application
Ongoing
Transitional periods apply for certain devices
References
- 1. EMA Guideline on quality requirements for drug-device combinations (EMA/CHMP/QWP/BWP/259165/2019)
- 2. Questions & Answers for applicants, marketing authorisation holders of medicinal products and notified bodies regarding medicines used in combination with medical devices (EMA/37991/2019 Rev.6)
- 3. Regulation (EU) 2017/745 on medical devices (MDR)
- 4. Directive 2001/83/EC relating to medicinal products for human use
- 5. MDCG 2022-5: Guidance on borderline between medical devices and medicinal products
Need faster answers? RegulatorySense delivers instant, authoritative guidance.
FAQ
What determines whether a DDC is regulated under MDR or pharmaceutical legislation?
When is a Notified Body Opinion required for integral DDCs?
What are the three conditions for Article 1(9) integral products?
What documentation goes in Module 3.2.R for DDCs?
Need Instant Clarity on DDC Requirements?
You've seen how complex the MDR Article 117 pathway can be. Our platform delivers instant, authoritative answers on DDC requirements.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Test Your Regulatory Knowledge → →5 questions · Personalized insights · Free