EMA Regulatory
Co-Packaged DDCs: Two Products, Two Sets of Rules
Quick Answer
Co-packaged drug-device combinations require the device to be independently CE marked under MDR, with its own labeling that follows MDR Annex I-completely separate from the medicinal product's pharmaceutical labeling. The MAH must provide evidence of device compliance with marketing authorization applications submitted after May 2021. Dual labeling means two parallel frameworks: Directive 2001/83/EC for the drug, MDR for the device. Devices up-classified under MDR benefit from a transition period until December 31, 2028.
Part 4 of a deep-dive series analyzing EMA's Questions and Answers on MDR/IVDR implementation. Part 1 covered Q1: DDC Classification. Part 3 covered Q3-4: Integral DDC Labeling. This post examines Q5-6-the dual-framework reality of co-packaged combinations.
The partner company's regulatory team calls at 4 PM on a Thursday. They've just been informed that the measuring spoon co-packaged with their oral solution now requires Notified Body certification.
Under the old Medical Devices Directive, the spoon was a Class I device-self-certified, minimal documentation, no NB involvement. Under MDR, it's been up-classified.
And now the marketing authorization holder has a question that neither team expected to face: what happens to the medicinal product if the co-packaged device can't get its certification in time?

The answer is more nuanced than either team expects. There are transition provisions. There are alternative labeling solutions for small devices. And there are responsibilities that need to be explicitly divided between the MAH and the device manufacturer-responsibilities that many partnership agreements never addressed because nobody anticipated the MDR would change the device's classification.
EMA's Q&A Questions 5 and 6 map this territory in detail-covering CE marking obligations, labeling separation, compliance evidence, and the specific provisions for devices that find themselves in a new regulatory class under MDR.
What Q5 and Q6 Actually Address
Why this matters: Co-packaged DDCs operate under dual regulation. Understanding where pharmaceutical and device requirements begin and end prevents compliance gaps that surface during assessment-or worse, post-market.
The EMA Q&A addresses co-packaged devices across Section 3 of the document (Q3.1 through Q3.4 in the numbering used by Revision 5). These questions cover the practical realities of a product configuration that many regulatory teams find more complex than integral DDCs-not because the rules are harder, but because there are two complete sets of rules running in parallel.
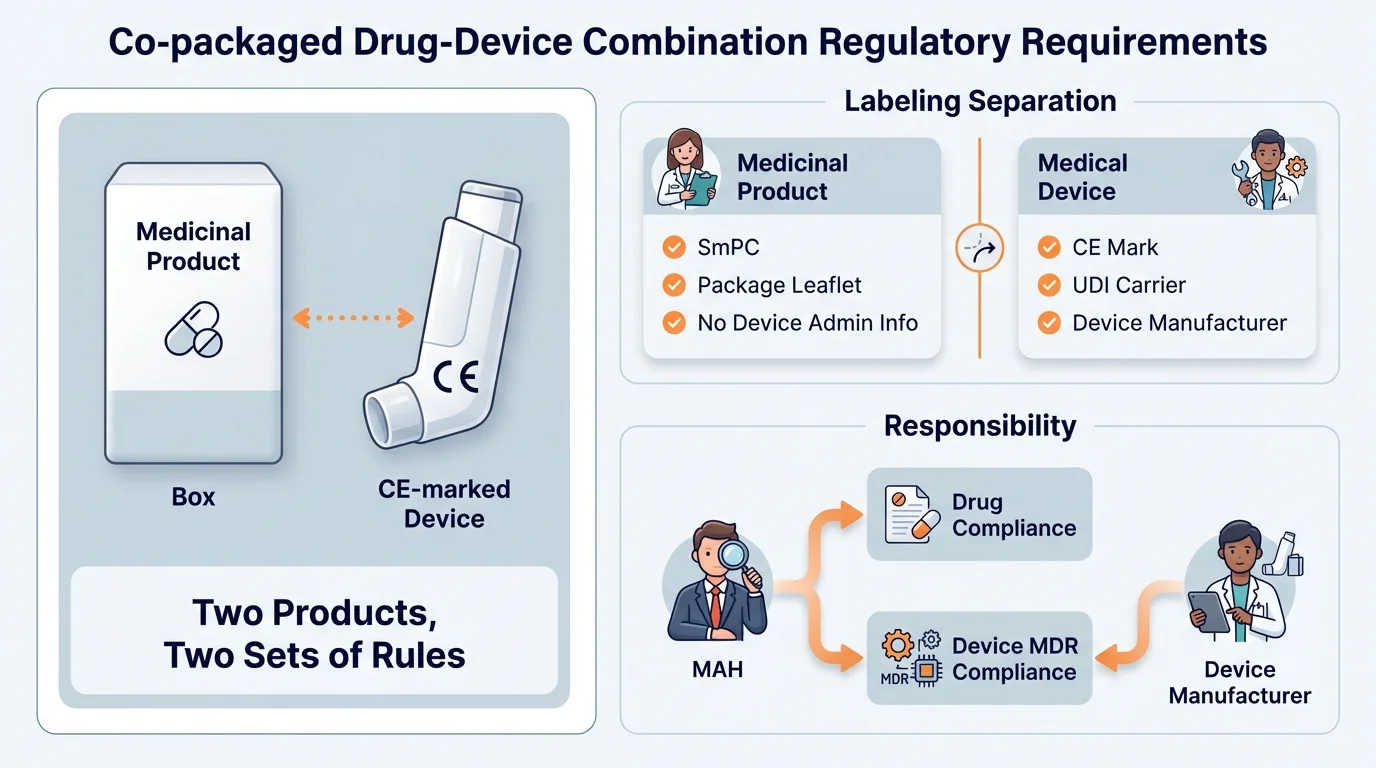
Unlike integral DDCs, where pharmaceutical labeling governs everything, co-packaged combinations are two distinct products placed in a single package. The medicinal product follows pharmaceutical legislation. The device follows MDR. Neither framework absorbs the other-they coexist, and the manufacturer (or MAH) must satisfy both simultaneously.
The CE Marking Obligation for Co-Packaged Devices
Why this matters: This is the fundamental difference from integral DDCs. Co-packaged devices must be independently CE marked-there's no exemption for being packaged with a medicinal product.
The Q&A is unambiguous: applicants for marketing authorizations that include a co-packaged medical device must ensure the device is CE marked in accordance with MDR to continue placing the product on the market. The device component undergoes its own conformity assessment, receives its own CE certificate (if required by classification), and carries its own CE marking.
This applies to everything from measuring spoons and oral syringes to spacers, inhalers, and nebulizers. If it's a medical device and it's co-packaged with a medicinal product, it needs CE marking under MDR.
For marketing authorization applications submitted after May 26, 2021, the MAH must provide evidence of device compliance. This evidence can take several forms:
Acceptable Evidence of Device Compliance
- • EU Declaration of Conformity - The device manufacturer's formal declaration that the device meets MDR requirements
- • EU Certificate from a Notified Body - For devices requiring NB assessment (Class Is, Im, Ir, IIa, IIb, III)
- • Other appropriate documentation - Demonstrating compliance with MDR Annex I GSPRs
Labeling: Two Products, Two Sets of Rules
Why this matters: The labeling separation is strict and frequently violated. Device administrative information must not leak into pharmaceutical product information, and pharmaceutical product information must not leak onto device labeling.
Co-Packaged DDC: Labeling Separation
| Labeling Element | Medicinal Product (Directive 2001/83/EC) | Co-Packaged Device (MDR 2017/745) |
|---|---|---|
| Regulatory Framework | Pharmaceutical legislation | Medical Device Regulation |
| CE Mark | Not included on drug product labeling | Required on device or device packaging |
| Manufacturer Details | MAH name and address only | Device manufacturer name and address |
| UDI | Not included on drug product labeling | Required on device per MDR |
| Notified Body Number | Not included on drug product labeling | Required if device was NB-certified |
| Instructions for Use | Safe use info integrated into SmPC and PL | Device-specific IFU if required by MDR Annex I Section 23 |
| Vigilance Reporting | Pharmacovigilance (EudraVigilance) | Device vigilance (EUDAMED when operational) |
The critical rule: The medicinal product's labeling (SmPC, Package Leaflet, outer packaging) follows Directive 2001/83/EC and should not include device administrative information-no device manufacturer name, no CE mark with NB number, no device symbols, no UDI, and no references to device vigilance reporting.
This separation protects both frameworks' integrity. When a patient or healthcare professional sees pharmaceutical product information, they should understand the product's regulatory status as a medicinal product. When they see the device, they should be able to identify it as a CE-marked device with its own regulatory pedigree. Mixing the two creates confusion about accountability, reporting obligations, and regulatory oversight.
Alternative Labeling for Small Devices
Why this matters: Many co-packaged devices (measuring spoons, oral syringes, droppers) are too small for direct MDR labeling. The Q&A provides three alternative solutions-but each has practical trade-offs that affect your packaging design and cost.
MDR labeling requirements-manufacturer name, CE mark, lot number, UDI carrier-are straightforward for devices with their own packaging. But many co-packaged devices are Class I or IIa, supplied without individual packaging, and physically too small to carry all required information. A measuring spoon can't accommodate a full MDR label.
The Q&A acknowledges this reality and provides three alternative solutions, each with distinct advantages:
Alternative Labeling Solutions for Small Co-Packaged Devices
- 1
Option A: Separate Device Leaflet
Include a separate, additional leaflet within the medicinal product's packaging specifically for the device's MDR administrative information. This results in two separate leaflets in the package, and a cross-reference between them is recommended.
- 2
Option B: Attached Leaflet
Attach the device administrative information leaflet to the medicinal product's Package Leaflet, forming a single folded component within the secondary packaging. This keeps all paper materials together while maintaining clear separation between drug and device information.
- 3
Option C: Fold-out Vignette or Sticker
Affix a fold-out vignette or sticker containing device-specific information directly onto the device or its packaging. The information must be indelible, legible, and comprehensible, and the adhesive must remain functional throughout the product's lifecycle.
Choosing the Right Alternative Labeling Solution
Question
What type of co-packaged device needs labeling?
Standard MDR labeling on device packaging - no alternative needed
If the device has its own packaging (even a simple wrapper), apply MDR labeling requirements directly to that packaging.
Use Option A, B, or C based on packaging constraints
Evaluate which solution best fits your packaging design, user experience, and regulatory documentation needs.
Direct marking preferred - supplement with leaflet if needed
MDR prefers direct marking on the device. If you can fit some required elements on the device, do so and supplement remaining information through one of the alternative options.
Standard MDR labeling on device packaging - no alternative needed
If the device has its own packaging (even a simple wrapper), apply MDR labeling requirements directly to that packaging.
Use Option A, B, or C based on packaging constraints
Evaluate which solution best fits your packaging design, user experience, and regulatory documentation needs.
Direct marking preferred - supplement with leaflet if needed
MDR prefers direct marking on the device. If you can fit some required elements on the device, do so and supplement remaining information through one of the alternative options.
Who Is Responsible for What
Why this matters: The division of responsibility between the MAH and the device manufacturer is where co-packaged DDC compliance most often breaks down. Without clear contractual agreements, changes to the device can blindside the MAH-and vice versa.
The Q&A establishes a clear principle: the MAH is responsible for ensuring that co-packaged devices comply with MDR before the combined product reaches the market. But how that responsibility is fulfilled depends on whether the MAH is also the device manufacturer.
Responsibility Division: MAH as Device Manufacturer vs Not
| Aspect | MAH Is Device Manufacturer | MAH Is Not Device Manufacturer |
|---|---|---|
| MDR Compliance Responsibility | MAH handles device compliance throughout lifecycle | Device manufacturer retains MDR compliance responsibility |
| Contact Details on Device | Only MAH details needed | Device manufacturer must be identified on device label and/or IFU |
| Change Management | Internal process-single entity controls both | Contractual agreements must govern how device changes are communicated to MAH |
| Vigilance Reporting | MAH handles both pharmacovigilance and device vigilance | Device manufacturer reports device incidents; MAH reports pharmaceutical adverse events |
| Post-Market Surveillance | MAH covers both drug and device PMS | Device manufacturer maintains device PMS system independently |
The practical imperative: If you're an MAH using a third-party device manufacturer, contractual agreements are not optional-they're essential. The Q&A emphasizes that agreements must ensure "appropriate communication and action regarding any changes to the device or its components." Without this, a device design change, a supplier change, or a recall can catch the MAH completely off guard.
When Your Co-Packaged Device Gets Up-Classified
Why this matters: MDR up-classified many previously Class I devices (measuring spoons, oral syringes, certain accessories). If your co-packaged device now requires Notified Body certification for the first time, there are transition provisions-but they have conditions and deadlines.
The transition from the Medical Devices Directive (MDD) to the Medical Device Regulation (MDR) reclassified several device types. Devices that were Class I under MDD-requiring only self-certification-may now be Class Is, Im, Ir, IIa, or even IIb under MDR, requiring Notified Body certification.
For co-packaged medical devices that are up-classified and require NB certification for the first time, the Q&A provides a clear pathway:
Transition Period: Until December 31, 2028
- • The device may remain on the market during the transition period
- • The device must continue to comply with MDD requirements
- • Conditions of MDR Article 120(3c) must be met
- • No significant changes to design or intended purpose are allowed
- • MDR requirements for PMS, vigilance, and economic operator registration apply immediately-not after transition
The last point is critical and often missed: transition provisions allow the device to remain on the market without MDR certification, but they do not exempt the device from MDR's post-market obligations. Post-market surveillance, market surveillance, vigilance reporting, and registration with economic operator databases must comply with MDR immediately-even while the device operates under MDD certification during the transition period.
FAQ
Must a co-packaged medical device be CE marked?
Can device administrative information appear on the medicinal product packaging?
What happens if a co-packaged device is up-classified under MDR and now needs Notified Body certification?
What evidence of device compliance is needed for marketing authorization applications?
Who is responsible for the co-packaged device's MDR compliance throughout its lifecycle?
Is Your Co-Packaged Device MDR-Compliant?
CE marking, dual labeling, manufacturer agreements-one gap in any of these creates an authorization risk. Two minutes to identify where your compliance might break.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Spot the Gaps →5 questions · Personalized insights · Free