EMA Regulatory
Ancillary Substance Consultation: The 210-Day Process That Gates Your CE Mark
Quick Answer
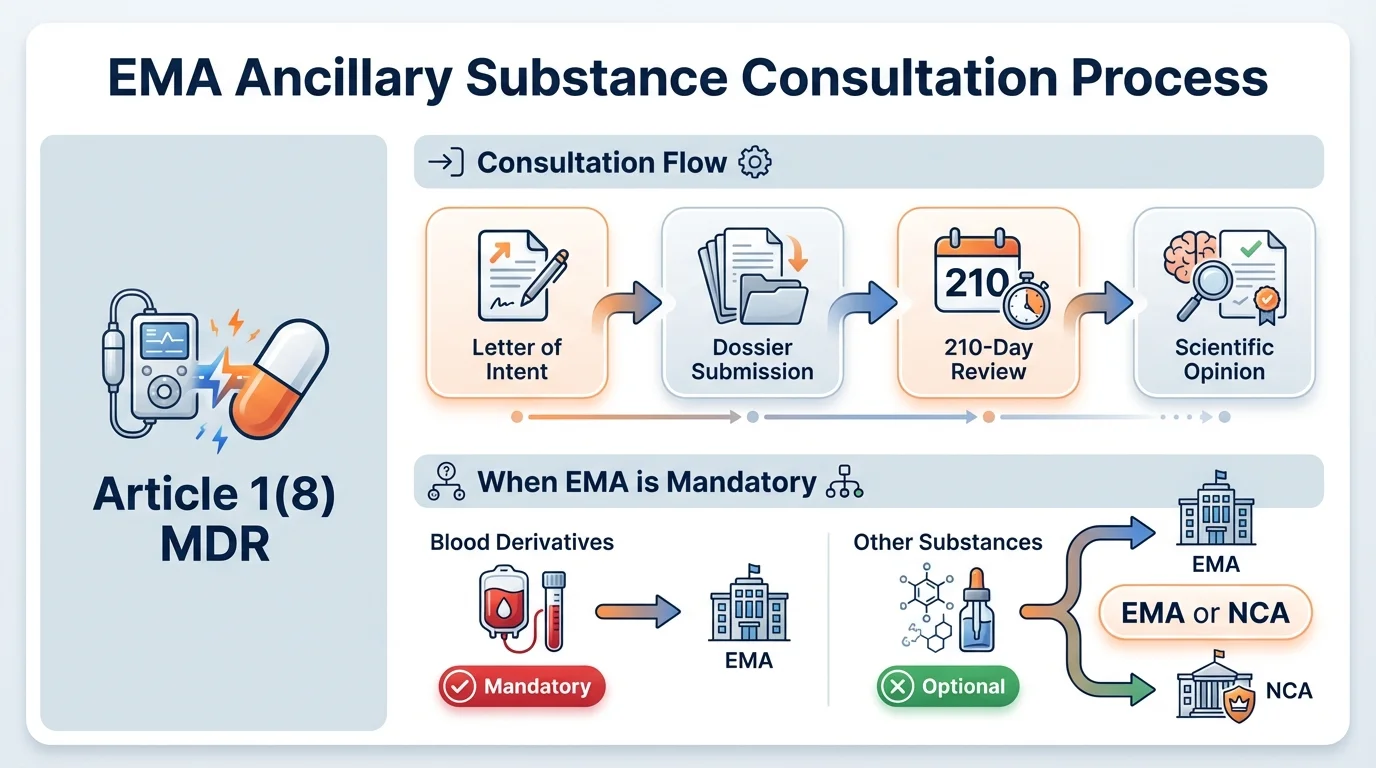
When a medical device incorporates an ancillary medicinal substance under Article 1(8) MDR, the Notified Body must obtain a scientific opinion on the substance's quality, safety, and usefulness before issuing a CE certificate. EMA consultation is mandatory for human blood derivatives and centrally authorized substances. For others, consultation can go to EMA or a national competent authority. The initial opinion takes up to 210 days. Changes to the substance post-certification trigger variation procedures analogous to pharmaceutical variations.
Part 5 of a deep-dive series analyzing EMA's Questions and Answers on MDR/IVDR implementation. Part 1 covered Q1: DDC Classification. Part 4 covered Q5-6: Co-Packaged Requirements. This post examines Q7-9-the consultation procedure for devices incorporating ancillary medicinal substances.
The gap analysis spreadsheet is 47 rows long. Each row represents a data element that EMA expects in an ancillary substance dossier-quality documentation, non-clinical data, clinical evaluation, manufacturing process details, justification of usefulness.
The regulatory affairs manager scrolls through the list again, comparing it against what the Notified Body has already compiled.
Fourteen rows are flagged red. Not because the data doesn't exist-it does, somewhere across three different departments and two contract manufacturers-but because nobody has assembled it into the format EMA actually requires.

And here's what makes this particularly consequential: until that scientific opinion arrives, the Notified Body cannot issue the CE certificate. The entire device certification is gated behind this consultation-and the 210-day assessment clock doesn't start until the dossier is validated as complete.
EMA's Q&A on the ancillary substance consultation procedure maps this territory in precise detail-from the letter of intent that starts the process, through the dossier requirements that determine whether your submission gets validated, to the variation procedures that govern every change you make to the substance after certification.
What Q7 Through Q9 Actually Address
Why this matters: The ancillary substance consultation is one of the least-understood bottlenecks in device certification. Many teams discover it exists only after starting the conformity assessment-adding months or years to their timeline.
The EMA's guidance on ancillary substance consultation spans two dedicated documents: the Q&A on the consultation procedure (EMA/144066/2021, Rev. 3) and the EMA Recommendation on procedural aspects and dossier requirements (EMA/CHMP/578661/2010, Rev. 2). Together, they cover the full lifecycle of the consultation-from initial intent through post-market changes.
The regulatory basis is Article 1(8) of MDR 2017/745 and Annex IX, Section 5.2. Any device that incorporates, as an integral part, a substance that would be considered a medicinal product if used separately-and where that substance has an action ancillary to the device-must undergo this consultation before certification.
The word "ancillary" is doing critical work here. It means the substance supports the device's primary function but doesn't define it. A drug-eluting stent, for example: the stent is the device, and the drug coating that prevents restenosis is the ancillary substance. The device's principal mode of action is mechanical. The drug's action is secondary-important, but ancillary.
Pro Tip
If the substance's action is the primary mode of action, the product is a medicinal product-not a device with an ancillary substance. The PMOA determination (covered in our guide on PMOA determination) must be resolved before the ancillary substance consultation applies.
When EMA Consultation Is Mandatory
Why this matters: Choosing the wrong authority-EMA vs national competent authority-doesn't just waste time. For blood derivatives and centrally authorized substances, EMA consultation isn't optional. Submitting to an NCA when EMA is required invalidates the entire assessment.
Which Authority Must Be Consulted?
Question
What type of ancillary substance does your device incorporate?
EMA consultation is MANDATORY - no alternative
Examples: Human albumin, fibrin sealants, thrombin-based coatings
EMA consultation is MANDATORY - no alternative
Centrally authorized medicinal products (biotech, orphan drugs, etc.)
Choice: EMA OR national competent authority (NCA)
Discretion of Notified Body in consultation with device manufacturer. EMA may be preferred where the substance was previously evaluated by EMA.
EMA consultation is MANDATORY - no alternative
Examples: Human albumin, fibrin sealants, thrombin-based coatings
EMA consultation is MANDATORY - no alternative
Centrally authorized medicinal products (biotech, orphan drugs, etc.)
Choice: EMA OR national competent authority (NCA)
Discretion of Notified Body in consultation with device manufacturer. EMA may be preferred where the substance was previously evaluated by EMA.
A practical consideration: Even when consultation with a national competent authority is permitted, many Notified Bodies prefer EMA-particularly when the ancillary substance is identical or closely related to a substance already assessed through the centralized procedure. The EMA's institutional memory on specific substances can streamline the assessment, because the same scientific teams that evaluated the standalone medicinal product may review the ancillary substance dossier.
One more critical distinction: if the EMA's scientific opinion is unfavorable for an ancillary human blood derivative, the Notified Body cannot issue the certificate. This is absolute-there's no provision for the Notified Body to override an unfavorable EMA opinion on blood-derived substances. For other ancillary substances, the Notified Body must give "due consideration" to EMA's opinion, which in practice means an unfavorable opinion is extremely difficult to overcome.
The Consultation Procedure Step by Step
Why this matters: The consultation procedure has specific sequencing requirements. Missing the letter of intent window or submitting before Rapporteurs are appointed creates delays that cascade through the entire certification timeline.
EMA Ancillary Substance Consultation: Complete Process
- 1
Letter of Intent
Submit to EMA at least 6 months before planned application date. Must be submitted at least 10 days before a CHMP meeting for inclusion on the agenda. Submit via EMA Service Desk under 'Pre-Submission Phase - Human'. You'll receive a Unique Product Identifier (UPI) for all future correspondence.
- 2
Rapporteur Appointment
EMA triggers appointment of Rapporteur, Co-Rapporteur, and Peer Reviewer (if applicable) by CHMP. Usually appointed at the next or subsequent CHMP meeting after receiving the letter of intent.
- 3
Dossier Submission
Submit complete application via eSubmission Gateway/Web Client (mandatory channel). Follow published CHMP assessment timetables for submission deadlines. No additional copies via CD/DVD or CESP.
- 4
Validation
EMA validates the application for completeness. Clock stops here if documentation is incomplete-the 210-day assessment period doesn't start until validation passes.
- 5
Scientific Assessment (210 Days)
CHMP assesses quality, safety, and usefulness of the ancillary substance. Takes into account the manufacturing process and usefulness of incorporating the substance into the device. May request additional information (clock stop).
- 6
Scientific Opinion Issued
EMA provides formal opinion to the Notified Body. If unfavorable (blood derivatives): NB cannot issue certificate. If unfavorable (other substances): NB must give due consideration. NB conveys final decision back to EMA.
The hidden timeline trap: Teams often plan around the 210-day assessment period, but the real timeline is longer. Add the 6-month pre-submission window for the letter of intent, time for Rapporteur appointment (1-2 CHMP meetings), and potential clock stops for information requests. A realistic end-to-end timeline from first contact to opinion is 12-18 months-not 7.
What Goes Into the Dossier
Why this matters: The dossier for ancillary substance consultation follows a structured format analogous to pharmaceutical submissions. Getting this wrong doesn't just delay validation-it signals to assessors that the applicant may not understand what's required, which colors their review of the substance itself.
The EMA Recommendation (Appendix 1) specifies five sections that mirror pharmaceutical dossier standards. The quality, safety, and usefulness of the substance are verified "by analogy with the methods specified in Annex I to Directive 2001/83/EC"-which means assessors evaluate the ancillary substance with the same rigor they'd apply to a standalone medicinal product, adjusted for the context of its incorporation into a device.
Ancillary Substance Dossier: Required Sections
- Section 1: Administrative information - Device description, manufacturer details, intended purpose, ancillary substance identification
- Section 2: Critical summaries - Quality, non-clinical, and clinical expert reports (or updates for variations)
- Section 3: Quality documentation - Drug substance and drug product quality data, manufacturing process, specifications, stability, sterilization impact
- Section 4: Non-clinical documentation - Toxicological assessment, pharmacodynamic and pharmacokinetic data relevant to device context
- Section 5: Clinical documentation - Clinical evaluation of the substance's benefit-risk profile within the device's intended purpose
The concept of "usefulness" is central to this assessment. Unlike a standalone medicinal product evaluation, the consultation specifically examines whether incorporating this substance into this device is justified. The Notified Body's report must explain why the substance is needed for the device's intended purpose, whether the substance achieves its intended action within the device context, and whether the inherent risks of the substance are justified by the benefit it provides.
This means quality and safety data alone aren't sufficient. You need to demonstrate that the substance should be there-that the device's performance depends on it, and that no safer alternative achieves the same result. Assessors will challenge weak usefulness justifications, and a substance that's safe but unnecessary won't receive a favorable opinion.
Post-Consultation: Managing Changes
Why this matters: Certification isn't the end. Every change to the ancillary substance-its manufacturing process, specifications, source, or formulation-triggers a variation procedure. Understanding which type of variation applies determines your timeline and documentation burden.
Changes to an ancillary medicinal substance are classified and evaluated by analogy to the pharmaceutical variations regulation (Commission Regulation (EC) No 1234/2008). This means the same framework that governs changes to marketed medicinal products applies to your ancillary substance-adapted for the device context.
Variation Types for Ancillary Substance Changes
| Aspect | Type IA Variation | Type IB Variation | Type II Variation |
|---|---|---|---|
| Nature of Change | Minor, no impact on quality/safety | Minor with potential quality/safety impact | Major change affecting quality, safety, or efficacy |
| Assessment Timeline | Notification-based | 30 days | 60 or 90 days |
| Examples | Administrative changes, minor specification tightening | Change in testing procedure, minor formulation adjustment | New supplier of substance, significant manufacturing process change |
| Submission Channel | eSubmission Gateway | eSubmission Gateway | eSubmission Gateway (follow published timetables) |
| Documentation | Cover letter, application form, relevant data | Cover letter, application form, relevant data | Cover letter, application form, updated quality/non-clinical/clinical summaries |
The critical obligation: Before any change is made to the ancillary substance-particularly its manufacturing process-the device manufacturer must inform the Notified Body. The Notified Body then seeks EMA's opinion to confirm that quality and safety remain unchanged. EMA must respond within 60 days for these change assessments. If the opinion is unfavorable, the Notified Body cannot issue the supplementary certificate.
Pro Tip
Devices with ancillary substances certified under the old Directives (93/42/EEC or 90/385/EEC) still need a new MDR-compliant consultation. The practical approach: leverage an upcoming variation-a change to the ancillary substance that triggers a follow-up consultation-to request an opinion in accordance with MDR. This avoids a completely new initial consultation while satisfying MDR requirements.
Timelines and Fees
Why this matters: The fee structure and realistic timelines are the two most-asked questions from device manufacturers encountering this procedure for the first time. Planning without accurate numbers leads to budget overruns and missed market access windows.
Realistic End-to-End Timeline
Month 0
Letter of Intent Submitted
Sent via EMA Service Desk at least 6 months before planned dossier submission. Includes UPI assignment.
Month 1-2
Rapporteur Appointment
CHMP appoints Rapporteur, Co-Rapporteur, and Peer Reviewer at next available meeting.
Month 6
Dossier Submission
Complete application submitted via eSubmission Gateway following CHMP assessment timetables.
Month 6-7
Validation
EMA validates completeness. Clock stops possible if data gaps identified.
Month 7-14
Scientific Assessment (210 Days)
CHMP evaluates quality, safety, and usefulness. Additional information requests may extend this period.
Month 14-18
Scientific Opinion & NB Decision
EMA issues opinion. Notified Body makes final certification decision based on opinion.
On fees: The medical device manufacturer-not the Notified Body-pays EMA's consultation fees directly. Fee amounts are published on the EMA Fees payable page under Annex IV, Section 7.1, updated with Regulation (EC) No 2024/568 effective January 2025. This catches some teams off guard because the Notified Body initiates the consultation, but the financial obligation sits with the manufacturer.
And here's what the published timelines don't capture: the preparation time. Assembling a complete dossier with quality, non-clinical, and clinical documentation-formatted to pharmaceutical standards-typically takes 6-12 months of internal preparation before the letter of intent is even submitted. The 210-day assessment period is only the middle chapter of a much longer story.
FAQ
When is EMA consultation mandatory for ancillary medicinal substances?
How long does the EMA consultation take for an ancillary medicinal substance?
What happens if EMA's scientific opinion on the ancillary substance is unfavorable?
Do devices with ancillary substances that were certified under the old Directives need a new EMA consultation under MDR?
Who pays the EMA consultation fees for ancillary substance assessment?
Would Your Ancillary Substance Dossier Pass EMA Review?
Missing data in Section 3 or Section 5 is the most common reason for clock stops during ancillary substance consultations. Two minutes to see where your documentation might have gaps.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Check Your Readiness →5 questions · Personalized insights · Free