EMA Regulatory
EU vs US Drug-Device Regulation: What Changes Everything
Quick Answer
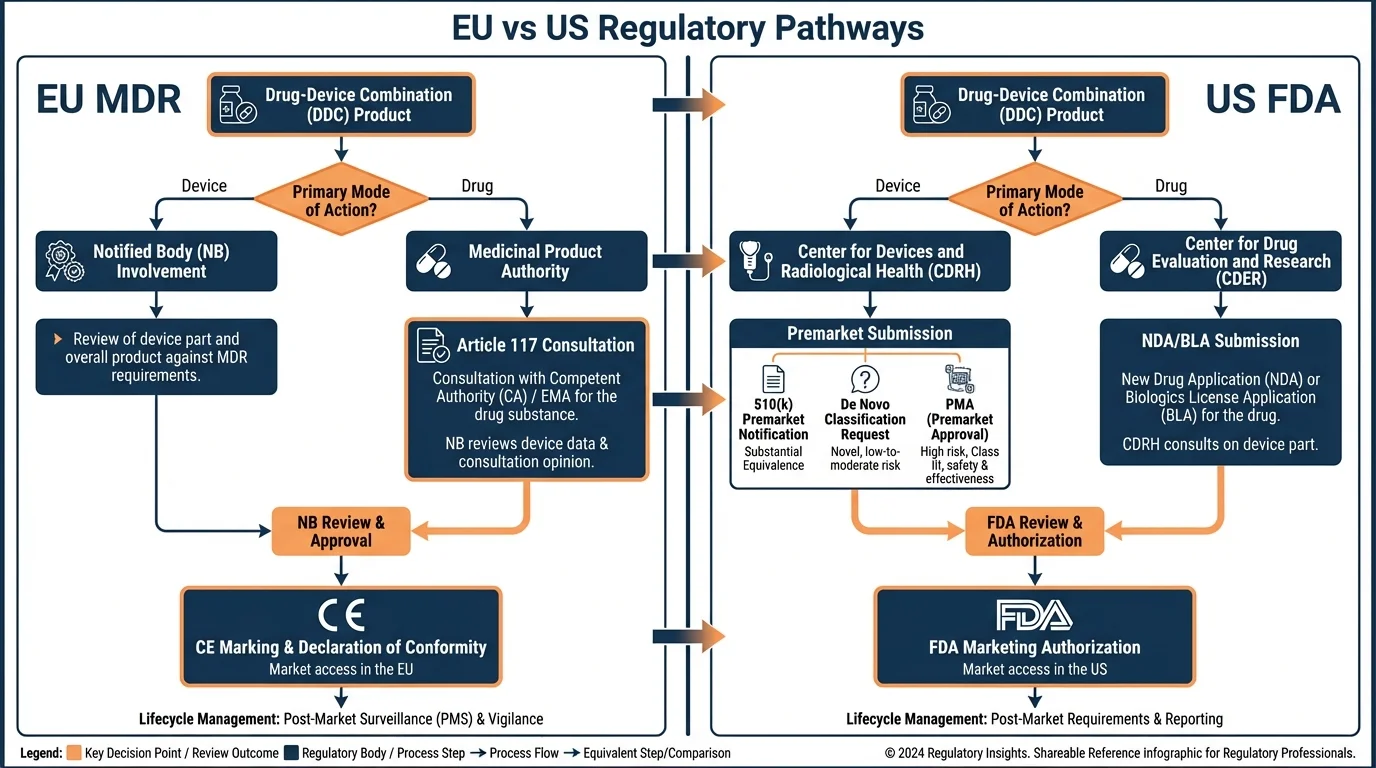
The EU requires dual assessment for drug-device combinations-a Notified Body evaluates device conformity under MDR while EMA assesses the medicinal product-whereas the US FDA uses a single application reviewed by one lead center determined by primary mode of action. This fundamental structural difference affects timelines, documentation, and regulatory strategy for every cross-border submission.
Over 60% of new biologic therapies now include a device component. Pre-filled syringes, autoinjectors, on-body delivery systems-the line between drug and device stopped being theoretical years ago.
His team had just secured FDA approval for a drug-autoinjector combination. Twelve months of review, one application, one center. Then the EU regulatory strategy landed on his desk, and what he saw looked like a different discipline entirely.

Two regulatory bodies. Two assessments instead of one. A Notified Body Opinion that didn't exist in the US pathway at all. And timelines that couldn't simply be run in parallel because the EU required the device assessment to feed into the marketing authorization application.
The difference between EU and US approaches to drug-device combinations isn't a matter of minor procedural variation. It's a structural divergence that reshapes how regulatory teams plan, resource, and execute their submissions from the earliest stages of development.
The Fundamental Difference: Single vs Dual Assessment
Why this matters: This single structural difference cascades into every other aspect of your regulatory strategy-timelines, resourcing, documentation, and stakeholder coordination.
The US FDA treats a drug-device combination product as one entity. You submit one application, to one center, and that center coordinates with other offices internally. The manufacturer interacts with a single regulatory body throughout.
The EU takes a fundamentally different approach. For drug-led combinations-where the medicinal product's action is principal-the device component must be assessed separately by a Notified Body before the overall marketing authorization can be granted by EMA or a national competent authority. This means two independent regulatory interactions, two sets of documentation requirements, and two assessment timelines that must be coordinated.
This dual assessment model was formalized under MDR Article 117, which amended pharmaceutical legislation to require that integral drug-device combinations include a Notified Body Opinion on device conformity with MDR Annex I General Safety and Performance Requirements (GSPRs) within the marketing authorization application.
The Coordination Challenge
The dual assessment doesn't mean duplicate review-the Notified Body evaluates device conformity against MDR Annex I, while the marketing authorization authority assesses overall safety and efficacy of the combined product. But coordinating these parallel tracks, and ensuring the Notified Body Opinion is ready when needed for the MAA dossier, is where many teams encounter timeline pressure.
EU MDR vs FDA: Framework Comparison
Why this matters: Understanding these structural differences early in development prevents costly mid-course corrections when preparing submissions for both markets.
EU MDR vs US FDA: Regulatory Framework Comparison
| Aspect | EU (MDR + Pharmaceutical Legislation) | US (FDA) |
|---|---|---|
| Definition Scope | 'Integral products' and 'co-packaged' DDCs under MDR Article 1(8)/(9) | Three types: single-entity, co-packaged, and cross-labeled (21 CFR Part 3) |
| Classification Basis | Principal vs ancillary action of medicinal substance | Primary Mode of Action (PMOA) per 21 CFR 3.2(m) |
| Drug-Led Assessment | Dual: NB Opinion on device + MA for product | Single application to CDER or CBER |
| Device-Led Assessment | CE marking via NB + medicinal authority consultation | Single application to CDRH (PMA, 510(k), or De Novo) |
| Post-Market Requirements | MDR PMS + pharmacovigilance obligations | GMP + QSR/CGMP + adverse event reporting |
| Lifecycle Changes | New NB Opinion may be required for device changes | Supplement to original application |
How Each System Classifies Combination Products
Why this matters: Classification determines your entire regulatory pathway-the authority you submit to, the documentation you prepare, and the timeline you plan around. Getting this right in both jurisdictions is the foundation of any cross-border strategy.
Both systems hinge on identifying the primary mechanism of action, but they operationalize this differently. The FDA assigns a lead review center through a formal PMOA determination process, while the EU distinguishes between products where the medicinal substance action is "principal" versus "ancillary" to determine which regulatory framework governs.
Which Regulatory Framework Applies?
Question
Is the medicinal product's action principal or ancillary?
EU: Marketing Authorization + NB Opinion (Article 117) | US: CDER/CBER lead (NDA/BLA)
The drug-device combination is regulated primarily as a medicinal product in both jurisdictions
EU: CE Marking via NB + Medicinal Authority Consultation | US: CDRH lead (PMA/510(k))
The product is regulated primarily as a medical device with drug-specific requirements
EU: Marketing Authorization + NB Opinion (Article 117) | US: CDER/CBER lead (NDA/BLA)
The drug-device combination is regulated primarily as a medicinal product in both jurisdictions
EU: CE Marking via NB + Medicinal Authority Consultation | US: CDRH lead (PMA/510(k))
The product is regulated primarily as a medical device with drug-specific requirements
One critical distinction: the FDA understands "combination products" more broadly than the EU. The FDA recognizes cross-labeled products-where a drug and device are sold separately but indicated for use together-as combination products. The EU does not treat separately marketed products as combination products under MDR, even when they're intended for combined use.
The EU Regulatory Pathway for Drug-Led DDCs
Why this matters: The EU pathway involves more stakeholders and coordination points than the FDA route. Understanding each step-and when to initiate it-is essential for realistic timeline planning.
EU Drug-Led DDC Approval Pathway
- 1
Select Notified Body
Identify MDR-designated NB with scope for your device type. Engage 12-18 months before MAA submission.
- 2
Prepare Device Documentation
Compile technical documentation demonstrating conformity with MDR Annex I GSPRs for the device component.
- 3
Obtain NB Opinion
NB conducts independent assessment and issues opinion report on device conformity.
- 4
Submit MAA with NB Opinion
Include NB Opinion in marketing authorization application to EMA (centralized) or NCA (national).
- 5
MA Assessment (210 days)
EMA evaluates overall product safety and efficacy. Clock stops at Day 120 and Day 180 for applicant responses.
- 6
Commission Decision
EU Commission grants marketing authorization within 67 days of positive CHMP opinion.
The Article 117 consultation procedure added a layer that didn't exist before MDR implementation. For integral drug-device combinations where the device component doesn't already have CE marking, the NB Opinion becomes a prerequisite for the MAA dossier-not a parallel activity that can be completed independently.
The FDA Regulatory Pathway
Why this matters: The FDA's single-application model creates different planning dynamics. Understanding these helps RA teams design parallel development strategies that account for both markets.
FDA's Office of Combination Products (OCP) determines the lead center based on PMOA. Once assigned, a single application covers the entire product. For drug-led combinations, this typically means an NDA or BLA to CDER or CBER, with device data included as part of the application rather than assessed separately.
FDA Advantages
- • Single point of contact throughout review
- • No separate device assessment coordination
- • Integrated review of drug-device interaction
- • Lifecycle changes via supplement to original application
FDA Considerations
- • PMOA disputes can delay center assignment
- • Cross-center review coordination is internal but not always seamless
- • Device quality system (QSR) requirements still apply
- • Pre-submission meetings recommended for novel combinations
Cross-Border Regulatory Strategy
Why this matters: Most drug-device combinations target both EU and US markets. A well-designed parallel strategy can save months compared to sequential submissions, but it requires early planning around both systems' requirements.
Cross-Border DDC Strategy Checklist
- Determine PMOA/principal action early-classification should align across both jurisdictions
- Engage EU Notified Body 12-18 months before planned MAA submission date
- Request FDA pre-submission meeting to confirm lead center assignment
- Design technical documentation to serve both MDR Annex I GSPR and FDA design control requirements
- Plan clinical program to generate evidence acceptable to both EMA and FDA
- Establish supplier data-sharing agreements early-EU NB Opinion requires detailed device documentation
- Map lifecycle change scenarios for both systems-EU may require new NB Opinion where FDA requires supplement
- Build separate but coordinated timelines-EU dual assessment typically adds 6-12 months vs FDA single application
The most common mistake in cross-border planning is assuming the EU pathway mirrors the US one with minor variations. Teams that have successfully navigated FDA review sometimes underestimate the coordination effort required for the EU's dual assessment model-particularly the lead time needed for Notified Body engagement and the dependency between NB Opinion timing and MAA readiness.
Practical Implications for RA Teams
Why this matters: Understanding structural differences at the regulatory framework level is necessary, but teams also need to know how these translate into day-to-day planning and resourcing decisions.
Resourcing Implications
EU submissions for drug-device combinations typically require specialized device regulatory expertise alongside pharmaceutical regulatory capabilities. This dual competency isn't always available within a single RA team. Organizations pursuing both markets need to plan for:
- • Device regulatory specialists familiar with MDR Annex I GSPRs
- • Pharmaceutical regulatory professionals managing the MAA
- • Project management to coordinate NB and EMA timelines
- • Quality system expertise spanning both MDR and pharmaceutical GMP
Documentation Strategy
Design your documentation architecture to serve both markets from the start. Key overlapping areas include:
- • Risk management files (ISO 14971 is recognized in both jurisdictions)
- • Design and development documentation (design controls overlap with MDR design requirements)
- • Clinical data (plan studies to meet both EMA and FDA evidence standards)
- • Biocompatibility testing (ISO 10993 series applies in both markets)
References
- 1. Regulation (EU) 2017/745 (MDR), Article 117 amending Directive 2001/83/EC
- 2. 21 CFR Part 3 - FDA Definition of Combination Products and PMOA determination
- 3. EMA Questions and Answers on implementation of MDR and IVDR (Revision 5, January 2025)
- 4. FDA Office of Combination Products - Guidance on Application Type for Combination Products
- 5. MDR Annex I - General Safety and Performance Requirements
- 6. Directive 2001/83/EC relating to medicinal products for human use, as amended by MDR Article 117
Need faster answers? RegulatorySense delivers instant, authoritative guidance with source citations.
FAQ
What is the main difference between EU and US regulation of drug-device combination products?
How does PMOA determination differ between the EU and US?
Do I need separate regulatory strategies for EU and US markets?
How does MDR Article 117 affect companies already approved in the US?
Navigating Both EU and US Pathways?
Two frameworks, two timelines, two sets of requirements. See how well you understand the differences that matter most.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Spot the Gaps →5 questions · Personalized insights · Free