EMA Regulatory
MDR Technical Documentation: Complete Guide to Annex II Requirements
Quick Answer
MDR Annex II technical documentation consists of six mandatory sections: device description, information supplied by manufacturer, design and manufacturing, GSPR compliance, risk management, and verification/validation. Each section must provide sufficient detail for Notified Body assessment of conformity.
You're preparing for your first MDR Notified Body audit. The auditor asks: "Show me how your technical documentation demonstrates GSPR compliance."
You hesitate. Your technical file is organized-you think-but is it organized the way Annex II requires? Will the auditor find the cross-references they need, or will you spend the next three hours hunting for documents?
This guide gives you the complete Annex II structure, section by section.
What Is Annex II Technical Documentation?
Annex II of the Medical Device Regulation (EU) 2017/745 defines the mandatory structure and content requirements for technical documentation that must accompany every medical device placed on the EU market.
The purpose of technical documentation is to:
"Allow assessment of the conformity of the device with the requirements of this Regulation."
Unlike the former Medical Device Directive (MDD), the MDR specifies the exact structure and content elements your technical file must contain. This isn't a suggestion-it's a regulatory requirement.
The Six Sections of Annex II
MDR Annex II organizes technical documentation into six distinct sections. Each builds on the previous, creating a comprehensive picture of your device from concept through market release.
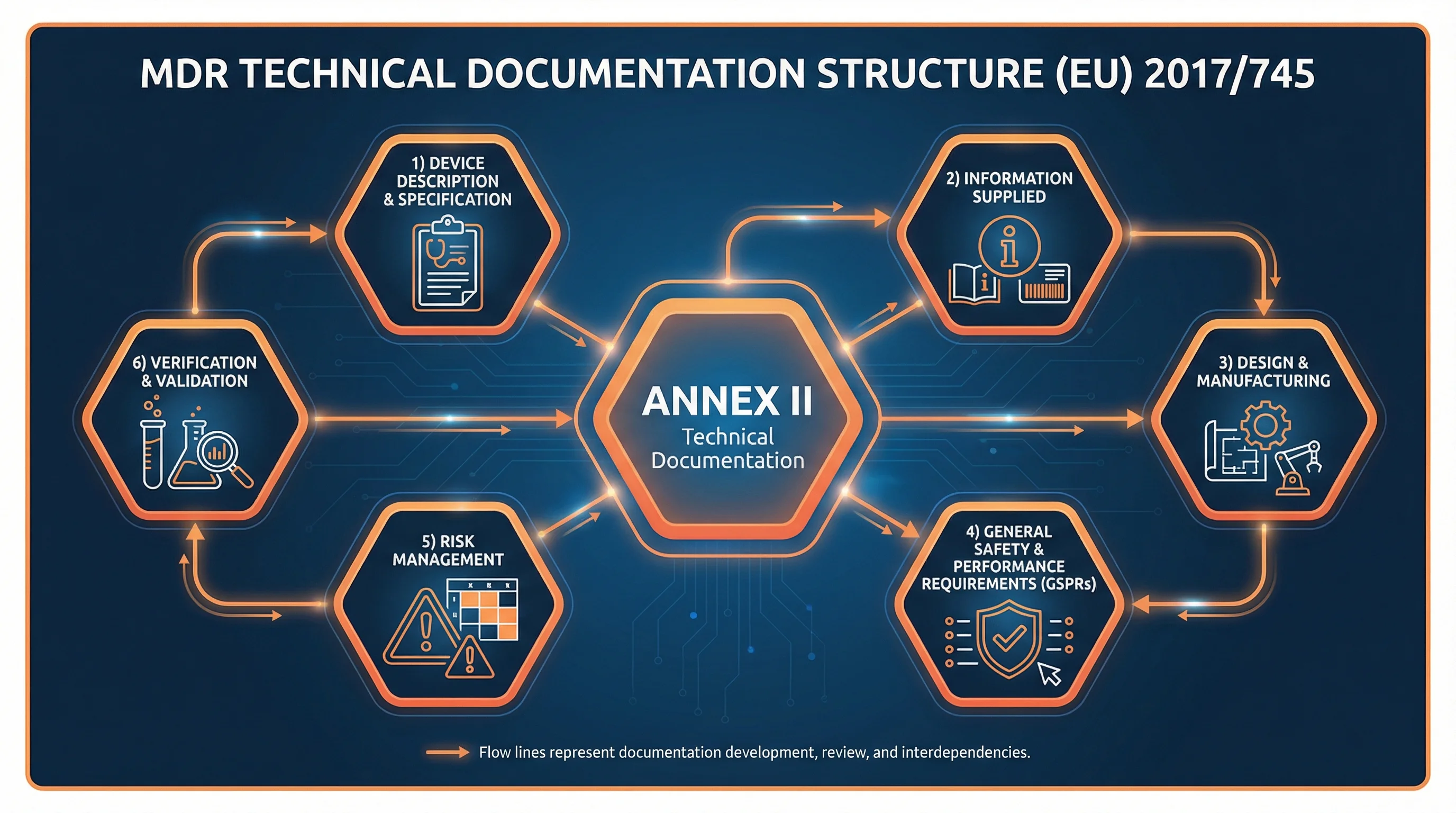
1
Device Description
What the device is and how it works
2
Information Supplied
Labels, IFU, and packaging
3
Design & Manufacturing
How the device is made
4
GSPRs
Safety and performance evidence
5
Risk Management
Benefit-risk and hazard analysis
6
Verification & Validation
Testing and clinical evidence
Section 1: Device Description and Specification
Section 1 establishes the foundation. A Notified Body reviewer reading this section should understand exactly what your device is, what it does, and who it's for.
Required Content
- • Product name and identification: Trade name, model numbers, Basic UDI-DI
- • Intended purpose: Clear, specific statement of what the device is designed to achieve
- • Intended users: Healthcare professionals, lay users, or both
- • Patient population: Age, condition, contraindications
- • Principle of operation: How the device achieves its intended purpose
- • Device classification: Class I, IIa, IIb, or III with classification rule justification
- • Variants and accessories: All configurations included under this technical documentation
- • Novel features: Any novel design, materials, or technologies
Best Practice
Include clear photographs, diagrams, and drawings. A reviewer should be able to visualize your device without ever seeing it in person. Annotate functional components and label all accessories.
Section 2: Information Supplied by Manufacturer
Section 2 covers all information that accompanies the device when it reaches the user-labels, instructions for use (IFU), and packaging.
Documentation Requirements
| Document Type | Key Requirements |
|---|---|
| Labels | UDI carrier, manufacturer info, lot/serial, CE mark, symbols per ISO 15223-1 |
| Instructions for Use | Intended purpose, contraindications, warnings, disposal, residual risks |
| Packaging | Sterility indication, storage conditions, opening instructions |
| Electronic IFU (eIFU) | Per Regulation (EU) 207/2012 if applicable |
All languages required for your target markets must be included. For EU-wide marketing, this typically means at least 24 official EU languages.
Section 3: Design and Manufacturing Information
Section 3 documents how your device is made. This includes design specifications, manufacturing processes, and supplier qualification.
Required Elements
Design Information
- • Complete design specifications and drawings
- • Design stages and design outputs
- • Bill of materials with specifications
- • Functional unit specifications
Manufacturing Information
- • Manufacturing site locations and activities
- • Process descriptions and flowcharts
- • Critical process parameters
- • Process validation documentation
- • Sterilization validation (if applicable)
Supplier Information
- • Critical supplier identification
- • Supplier qualification records
- • Supplier agreements and specifications
- • Incoming inspection requirements
Section 4: General Safety and Performance Requirements
This is the heart of your technical documentation. Section 4 demonstrates how your device meets each applicable General Safety and Performance Requirement (GSPR) from MDR Annex I.
The GSPR Checklist
The cornerstone of Section 4 is your GSPR checklist. This document maps each Annex I requirement to your evidence of compliance.
| GSPR | Applicable? | Standard/Method | Evidence Location |
|---|---|---|---|
| 1. Safety and performance | Yes | ISO 14971, IEC 62366-1 | RA-001, UE-001 |
| 10. Chemical properties | Yes | ISO 10993-18 | BC-003 |
| 17. Radiation | No - N/A | - | Justification in RA-001 |
Common Audit Finding
Non-applicable GSPRs must include a justification explaining why they don't apply. Simply marking "N/A" without explanation is a frequent nonconformity.
Harmonized Standards
Using harmonized standards provides a presumption of conformity with the corresponding GSPRs. Document which standards you've applied and confirm you've met all relevant clauses.
If you deviate from a harmonized standard, document your alternative approach and demonstrate equivalence or superiority.
Section 5: Benefit-Risk Analysis and Risk Management
Section 5 documents your complete risk management process per ISO 14971, plus your overall benefit-risk determination.
Required Documentation
- • Risk Management Plan: Scope, criteria, team responsibilities, review milestones
- • Risk Analysis: Hazard identification, hazardous situations, harm estimation
- • Risk Evaluation: Risk acceptability decisions per your criteria
- • Risk Control: Control measures, verification of effectiveness, residual risk
- • Overall Residual Risk Evaluation: Aggregate assessment of all residual risks
- • Benefit-Risk Determination: Benefits outweigh residual risks
- • Risk Management Report: Summary confirming plan completion
Your risk management file must be a living document, updated throughout the product lifecycle as new hazards are identified through post-market surveillance.
Section 6: Product Verification and Validation
Section 6 provides the evidence that your device works as intended. This includes pre-clinical testing, biocompatibility, software verification, and clinical evaluation.
Key Documentation Areas
Pre-Clinical Testing
- • Bench testing reports
- • Biocompatibility (ISO 10993 series)
- • Electrical safety (IEC 60601-1)
- • EMC testing (IEC 60601-1-2)
- • Sterilization validation
- • Shelf-life/stability studies
- • Packaging validation
Clinical Evidence
- • Clinical Evaluation Report (CER)
- • Clinical investigation reports
- • Literature review
- • Equivalence analysis (if applicable)
- • PMCF plan
- • SSCP (Class III/implantables)
Software Documentation (if applicable)
For devices containing software, Section 6 must include:
- • Software development lifecycle documentation per IEC 62304
- • Software requirements and architecture
- • Verification and validation test results
- • Cybersecurity risk assessment
- • Software of Unknown Provenance (SOUP) analysis
Practical Steps for Building Your Technical File
Building MDR-compliant technical documentation is a significant undertaking. Here's a practical approach:
Start with Gap Analysis
Compare your existing documentation against Annex II requirements. Identify what exists, what needs updating, and what's missing entirely.
Establish Document Structure
Create a master document index that maps to Annex II sections. Use consistent naming conventions and version control.
Build the GSPR Checklist First
The GSPR checklist drives everything else. Determine applicable requirements before generating evidence.
Cross-Reference Extensively
Auditors navigate your documentation through cross-references. Every claim should point to supporting evidence; every test should trace back to a requirement.
Plan for Updates
Technical documentation is never "done." Establish processes for incorporating design changes, post-market data, and regulatory updates.
Key Success Factor
The best technical files tell a coherent story. A reviewer should be able to follow the thread from intended purpose through risk identification through design controls through testing to clinical evidence-all supporting the same claims.
FAQ
What's the difference between MDR Annex II and Annex III?
Do I need to demonstrate compliance with ALL GSPRs in Annex I?
When is a Summary of Safety and Clinical Performance (SSCP) required?
What format should technical documentation follow?
Need Instant Clarity on Technical Documentation?
You've seen how detailed Annex II requirements can be. Our platform delivers instant, authoritative answers on technical documentation structure and content.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Get Regulatory Guidance → →5 questions · Personalized insights · Free