EMA Regulatory
EU MDR Post-Market Surveillance: The Complete Guide to PMS, PSUR, and Vigilance
Quick Answer
Post-market surveillance under EU MDR requires all manufacturers to implement a documented PMS system (Article 83) based on a PMS plan (Article 84). Class I devices need PMS Reports; Class IIa/IIb/III require Periodic Safety Update Reports (PSURs) at defined intervals. The system must actively collect safety and performance data throughout the device's lifetime.
You're preparing for a Notified Body audit. The assessor asks: "Show me how your PMS system feeds into your clinical evaluation updates."
You pause. You have a PMS plan. You have PSURs. But can you trace the data flow from a complaint to a clinical evaluation update to a label change? The MDR expects you to-and the consequences of a disconnected system are audit findings that delay your certification.
This guide gives you the complete framework that auditors expect to see.
What Is Post-Market Surveillance?
Why this matters: PMS isn't a compliance checkbox-it's the foundation for every safety decision you'll make after your device reaches the market.
Post-market surveillance is a proactive and systematic process for collecting, analyzing, and acting on data about your device once it's on the market. Under the MDR, this isn't optional or passive-manufacturers must actively gather information throughout the device's entire lifetime.
Article 83 of the MDR defines PMS as a system that must be:
"suited to actively and systematically gathering, recording and analysing relevant data on the quality, performance and safety of a device throughout its entire lifetime, and to drawing the necessary conclusions and to determining, implementing and monitoring any preventive and corrective actions."
The key word is "proactive." Unlike the old Medical Device Directive (MDD), where PMS was often reactive, the MDR expects you to seek out data rather than wait for it to find you.
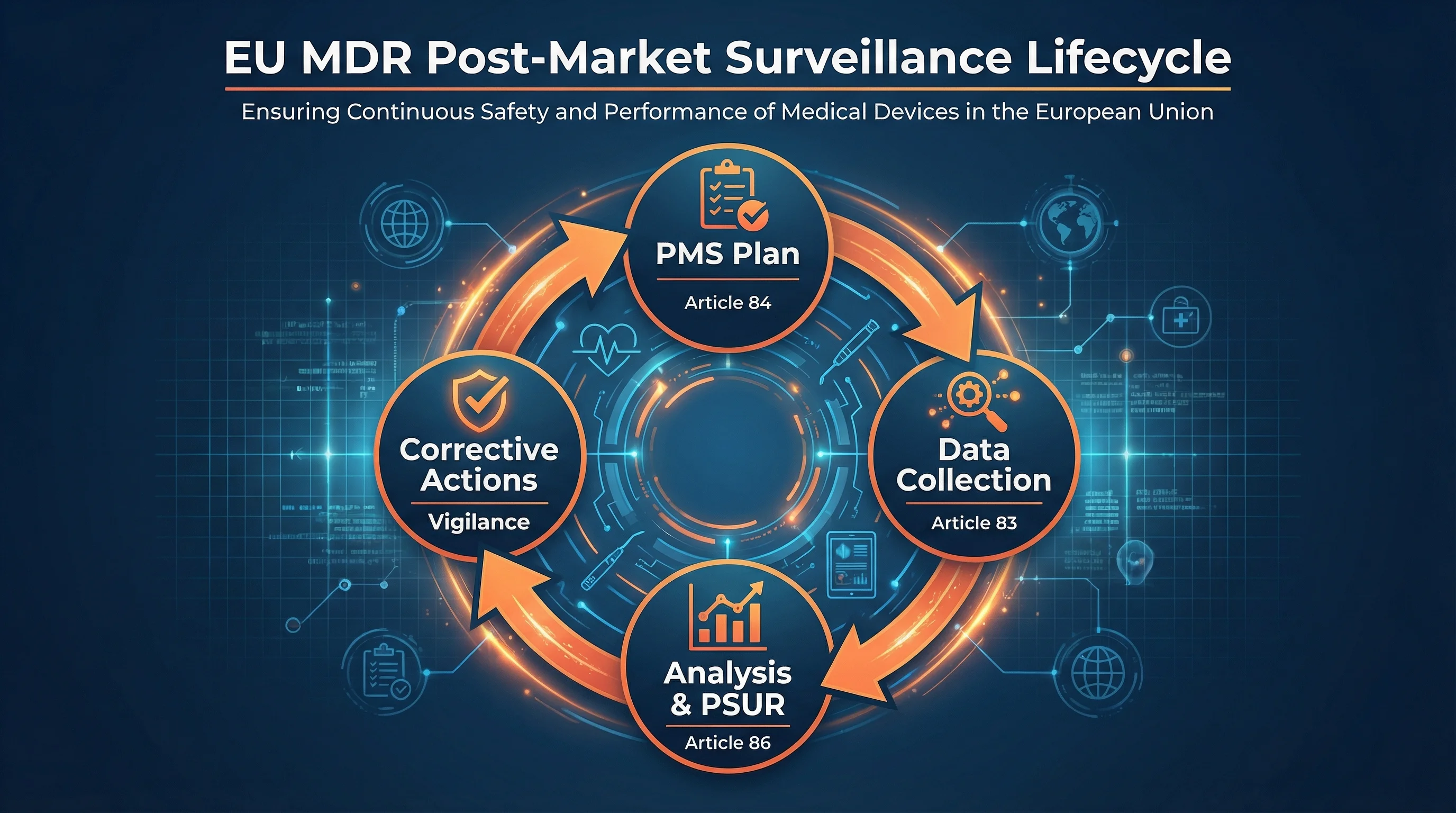
The PMS Lifecycle Under EU MDR
- 1
PMS Plan
Document data sources, collection methods, and analysis procedures (Article 84, Annex III)
- 2
Data Collection
Actively gather complaints, vigilance data, literature, registries, and user feedback (Article 83)
- 3
Analysis & Reporting
Evaluate data, update benefit-risk, prepare PMS Report or PSUR (Articles 85-86)
- 4
Corrective Actions
Implement CAPAs, update clinical evaluation, trigger vigilance if needed (Articles 87-92)
The PMS Legal Framework (Articles 83-86)
Why this matters: These four articles form the backbone of your PMS obligations. Understanding their hierarchy prevents documentation gaps that trigger audit findings.
The MDR establishes PMS requirements across four interconnected articles:
Article 83 - PMS System
Sets the overarching obligation: every manufacturer must establish, document, implement, and maintain a PMS system that is proportionate to the device's risk class and type.
Key point: Applies to ALL manufacturers, regardless of device class-including Class I.
Article 84 - PMS Plan
Requires the PMS system to be based on a documented PMS plan. This plan must be part of the technical documentation (Annex II) for all devices except custom-made devices.
Key point: The plan is a living document that must address the elements in Annex III, Section 1.1.
Article 85 - PMS Report
Manufacturers of Class I devices must prepare a PMS Report summarizing PMS data, conclusions, and any preventive/corrective actions taken.
Key point: Updated when necessary; made available to competent authorities upon request.
Article 86 - Periodic Safety Update Report (PSUR)
Manufacturers of Class IIa, IIb, and III devices must prepare PSURs that include benefit-risk conclusions, PMCF findings, sales volume, and population estimates.
Key point: PSURs have mandatory update frequencies and Notified Body submission requirements.
PMS Plan Requirements (Annex III)
Why this matters: Annex III Section 1.1 is what auditors check first. A complete PMS plan prevents the most common certification delays.
Your PMS plan must address specific elements required by Annex III. This isn't a template you fill in once-it's a system design document that explains how you'll actually operate.
PMS Plan Required Elements (Annex III Section 1.1)
- 1 Proactive data collection procedures - complaints, user feedback, literature, registries
- 2 Methods for analyzing collected data - statistical methods, trending thresholds
- 3 Indicators and threshold values for benefit-risk reassessment - triggers for action
- 4 Methods for investigating complaints and market feedback - timelines, escalation paths
- 5 Methods for managing events subject to trend reporting - Article 88 thresholds
- 6 Methods for communication with competent authorities and NBs - reporting formats, contacts
- 7 Procedures for FSCA and recalls - decision criteria, implementation steps
- 8 Reference to PMCF plan - or justification if PMCF not applicable
- 9 Procedures for keeping technical documentation updated - IFU, clinical evaluation, labeling
Pro tip: Your PMS plan should reference your Quality Management System (QMS) procedures rather than duplicating them. Auditors want to see integration, not standalone documents.
PSUR Timeline by Device Class
Why this matters: Missing a PSUR deadline can trigger a certificate suspension. Know your class-specific requirements.
Which Report Do You Need?
Question
What is your device's risk class?
PMS Report (Article 85)
Update as necessary. No fixed frequency. Make available to CA upon request.
PSUR every 2 years
Make available to NB and CA upon request.
PSUR annually
Submit to NB as part of surveillance. NB evaluates and adds to EUDAMED.
PMS Report (Article 85)
Update as necessary. No fixed frequency. Make available to CA upon request.
PSUR every 2 years
Make available to NB and CA upon request.
PSUR annually
Submit to NB as part of surveillance. NB evaluates and adds to EUDAMED.
PMS Report vs PSUR: Key Differences
| Aspect | PMS Report (Article 85) | PSUR (Article 86) |
|---|---|---|
| Applies to | Class I devices | Class IIa, IIb, III devices |
| Update frequency | When necessary | Every 1-2 years (class-dependent) |
| Content | PMS data summary, conclusions, CAPAs | Benefit-risk determination, PMCF findings, sales volume, population estimate |
| Submission | Available on request | NB submission required (IIb/III) |
| EUDAMED | Not uploaded | NB uploads to EUDAMED (when functional) |
Vigilance and Serious Incident Reporting
Why this matters: Vigilance failures have the most severe consequences-from certificate suspension to criminal liability. Get reporting timelines right.
Vigilance is the reactive component of PMS-what you do when things go wrong. Under MDR Article 87, manufacturers must report serious incidents and Field Safety Corrective Actions (FSCAs) to competent authorities.
What Qualifies as a "Serious Incident"?
Any incident that directly or indirectly led, might have led, or might lead to:
- Death of a patient, user, or other person
- Serious deterioration in the state of health of a patient, user, or other person
- A serious public health threat
Serious Public Health Threat
Report within 2 days
Death or Serious Deterioration
Report within 10 days (15 days if more time needed)
Other Reportable Incidents
Report within 15 days
Field Safety Corrective Actions (FSCAs)
When you implement a corrective action to reduce risk from devices already on the market, you must:
- Report the FSCA to competent authorities (typically before or concurrent with implementation)
- Issue a Field Safety Notice (FSN) to users and, where relevant, patients
- Document the rationale, scope, and implementation verification
- Follow up with a final report once the FSCA is complete
PMCF: The Clinical Component of PMS
Why this matters: PMCF bridges your clinical evaluation and PMS system. Without it, you can't demonstrate ongoing benefit-risk acceptability.
Post-Market Clinical Follow-up (PMCF) is a continuous process that proactively collects and evaluates clinical data from devices in use. It's defined in MDR Annex XIV Part B and must be part of your clinical evaluation.
PMCF can use both:
Active PMCF Methods
- PMCF studies (surveys, registries, prospective studies)
- Questionnaires to healthcare professionals
- Targeted follow-up of specific patient populations
- Analysis of real-world evidence databases
Passive PMCF Methods
- Ongoing literature surveillance
- Analysis of complaints and vigilance data
- Review of registry data
- Monitoring of comparable devices
Can You Justify Not Doing PMCF?
Yes, but only if you can demonstrate that adequate clinical evidence already exists and additional PMCF wouldn't add value. This justification must be:
- Documented in the clinical evaluation report
- Based on the device's clinical history and literature coverage
- Approved by the Notified Body (for Class IIa+)
- Reassessed whenever new information emerges
Practical Steps for Implementation
For New Device Launches
- 1. Design your PMS plan early - Don't wait until conformity assessment. Integrate PMS planning into design and development.
- 2. Define data sources - Identify proactive sources (registries, user surveys, literature databases) not just reactive ones (complaints).
- 3. Set quantitative thresholds - Define what complaint rate or adverse event frequency triggers a benefit-risk review.
- 4. Align with clinical evaluation - Your PMCF plan should fill gaps identified in the clinical evaluation.
- 5. Train your team - Ensure everyone who handles complaints or user feedback knows the vigilance reporting criteria.
For Existing Devices (MDD to MDR Transition)
- 1. Gap analysis - Compare your current PMS system against MDR Annex III requirements.
- 2. Retrospective data - Compile PMS data from your MDD period for your first PSUR.
- 3. PMCF justification - If you didn't have formal PMCF, prepare the justification or plan new activities.
- 4. EUDAMED readiness - Prepare for PSUR uploads when EUDAMED vigilance module goes live.
Integration with QMS
Your PMS system should be integrated with-not separate from-your Quality Management System:
- Link PMS findings to CAPA procedures
- Connect clinical evaluation updates to design changes
- Align vigilance reporting with document control
- Include PMS review in management review meetings
- Test your understanding of these integration points
Key MDCG Guidance Documents
MDCG 2022-21
Guidance on PSUR according to MDR
MDCG 2020-7
PMCF Plan Template
MDCG 2020-6
Guidance on Clinical Evaluation - PMCF
MDCG 2019-16
Cybersecurity Guidance (includes PMS for software)
References
- 1. Regulation (EU) 2017/745 on medical devices (MDR) - Articles 83-92, Annex III
- 2. MDCG 2022-21: Guidance on Periodic Safety Update Report (PSUR)
- 3. MDCG 2020-6: Guidance on sufficient clinical evidence for legacy devices
- 4. MDCG 2020-7: PMCF Plan Template
- 5. MDCG 2019-16 Rev.1: Guidance on Cybersecurity for medical devices
Need faster answers? RegulatorySense delivers instant, authoritative guidance.
FAQ
What is the difference between a PMS Report and a PSUR?
How often must a PSUR be updated under MDR?
When is PMCF required, and can it be waived?
What must be reported as a 'serious incident' under MDR vigilance?
Struggling with PMS Documentation Requirements?
You've seen how interconnected the PMS framework can be. Our platform delivers instant, authoritative answers on PMS, PSUR, and vigilance requirements.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Test Your PMS Knowledge →5 questions · Personalized insights · Free