EMA Regulatory
MDR 2017/745 for Drug-Device Combinations: What Every RA Professional Must Know
Quick Answer
The Medical Device Regulation (EU) 2017/745 governs drug-device combinations based on principal mode of action. If the device's action is principal, MDR applies fully. If the medicinal product's action is principal, the product follows pharmaceutical legislation but the device component must still comply with MDR Annex I GSPRs-demonstrated via EU certificate, Declaration of Conformity, or Notified Body Opinion.
Three weeks before the submission deadline, his technical reviewer flags an issue: "The device component section references MDD. Shouldn't this be MDR now?"
He checks the timeline. The MAA was drafted when MDD certificates were still valid.
Now the transitional period has shifted, and half his GSPR references are pointing to the wrong regulation.

The gap that costs months: understanding exactly how MDR 2017/745 applies to your combination product-not just knowing it exists, but knowing which provisions trigger which requirements.
Breaking it down: the complete framework for MDR compliance in drug-device combinations.
MDR Scope: Where Drug-Device Combinations Fit
Why this matters: Misunderstanding MDR scope leads to either over-engineering your submission or missing critical requirements entirely.
The Medical Device Regulation (EU) 2017/745 replaced the Medical Device Directive (93/42/EEC) as the primary legislation governing medical devices in the European Union. But its impact extends well beyond standalone devices.
For drug-device combinations, MDR creates a dual framework:
When MDR Applies Directly
The device's mode of action is principal
- • Device incorporating ancillary medicinal substance
- • Full MDR conformity assessment required
- • CE marking on the combination product
- • EMA/NCA scientific opinion on the substance
When MDR Applies Indirectly
The medicinal product's action is principal
- • Product regulated as medicinal product
- • Device component must meet relevant GSPRs
- • Evidence via EU certificate, DoC, or NB Opinion
- • Article 117 pathway applies
The key insight: even when your combination product is regulated primarily as a medicinal product, MDR doesn't disappear. It establishes the safety and performance baseline your device component must meet.
The Two Pathways: Device-Led vs Medicine-Led
Why this matters: The principal mode of action (PMOA) determines your entire regulatory strategy. Getting this wrong means rebuilding your dossier from scratch.
MDR distinguishes between two types of integral drug-device combinations based on which action is principal:
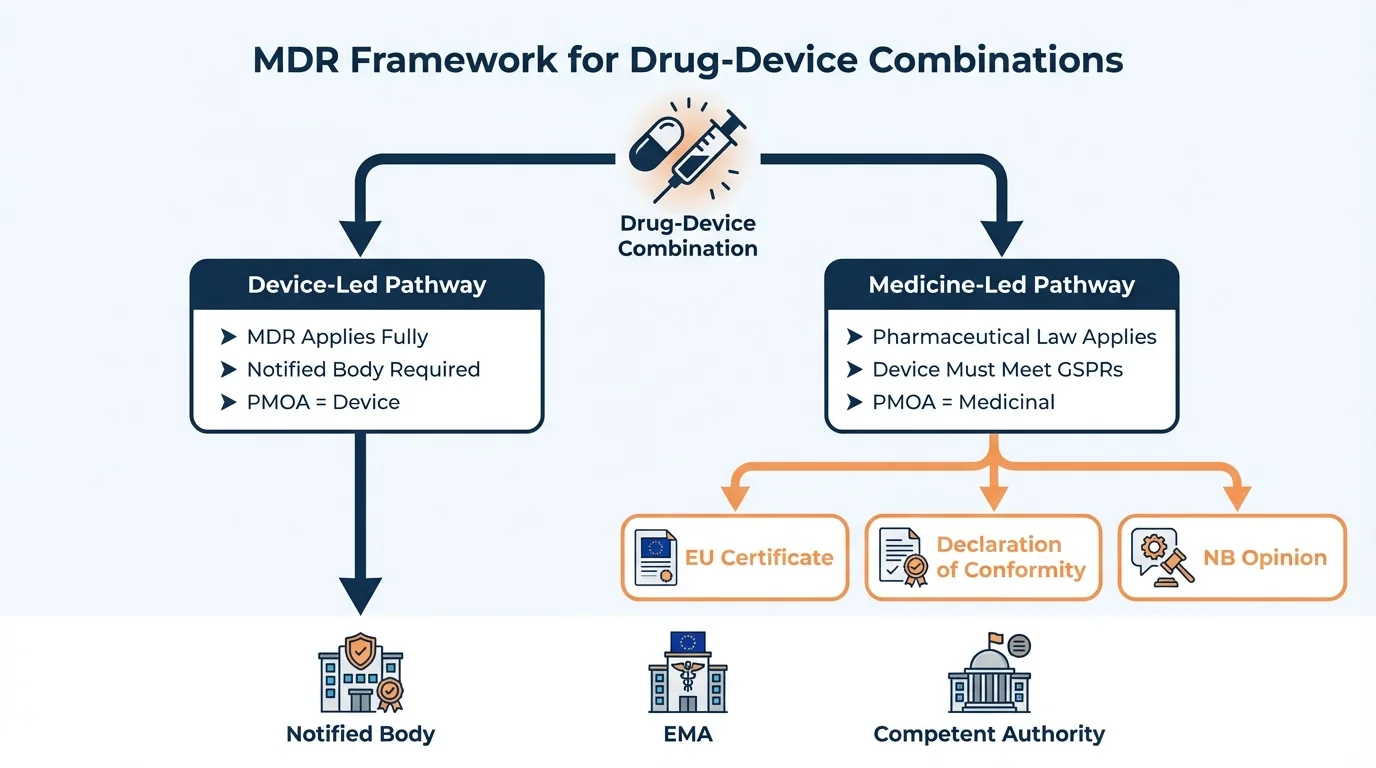
How to Use This Decision Framework: Start with the primary intended purpose of your product. If the therapeutic benefit comes primarily from the medicinal substance, you're on the medicine-led pathway. If the device function delivers the primary benefit (with the substance supporting), you're on the device-led pathway.
Question
What is the principal mode of action of your combination product?
Medicine-led pathway: Regulated under Directive 2001/83/EC. Device component must meet MDR Annex I GSPRs. Submit via Article 117.
Device-led pathway: Regulated under MDR 2017/745. Substance has ancillary action. Requires EMA/NCA scientific opinion before CE marking.
Medicine-led pathway: Regulated under Directive 2001/83/EC. Device component must meet MDR Annex I GSPRs. Submit via Article 117.
Device-led pathway: Regulated under MDR 2017/745. Substance has ancillary action. Requires EMA/NCA scientific opinion before CE marking.
The PMOA determination isn't always obvious. For borderline cases, MDCG 2022-5 provides detailed guidance, and national competent authorities can issue formal classification advice.
Medicine-Led DDCs: Article 1(8) and 1(9)
When the medicinal substance has the principal action, MDR Article 1(8) and 1(9) define how the device component is handled:
Article 1(8): Device with Ancillary Substance
A medical device that incorporates a substance that, if used separately, would be a medicinal product with principal action.
Regulatory outcome: The integral product is governed by medicinal product legislation (Directive 2001/83/EC or Regulation 726/2004).
Article 1(9): Device for Administering Medicinal Product
A device intended to administer a medicinal product where they form a single integral product, intended exclusively for that combination, and not reusable.
Examples: Pre-filled syringes, autoinjectors, pre-charged inhalers, transdermal patches with reservoir.
Device-Led DDCs: Ancillary Medicinal Substance
When the device's mode of action is principal, the product is regulated fully under MDR. The medicinal substance is considered "ancillary"-supporting the device's primary function.
Before the Notified Body can issue a CE certificate, they must obtain a scientific opinion from either:
- A National Competent Authority (NCA) for medicinal products, OR
- The European Medicines Agency (EMA)
This opinion addresses the quality and safety of the substance, including the benefit/risk of incorporating it into the device.
General Safety and Performance Requirements (GSPRs)
Why this matters: GSPRs are the backbone of MDR compliance. For combination products, you need to demonstrate compliance even when regulated primarily as a medicinal product.
MDR Annex I contains the General Safety and Performance Requirements-the essential standards every medical device must meet. For combination products, the relevant GSPRs depend on the device component's function and risk profile.
GSPR Categories Most Relevant to DDCs:
Key GSPR Categories for Drug-Device Combinations
- Chapter I: General requirements (risk management, design for safety)
- Chapter II: Design and manufacture (materials, contamination, interaction with substances)
- Chapter III: Information supplied with the device (labeling, IFU)
- Section 12: Devices incorporating substances (specific to DDCs)
- Section 14: Devices with measuring function (if applicable)
- Section 22: Software (if device includes software components)
Section 12: The Critical GSPR for DDCs
Annex I, Section 12 specifically addresses devices incorporating substances that, if used separately, would be considered medicinal products. Key requirements include:
| Requirement | What It Means |
|---|---|
| Quality verification | Substance must meet quality standards analogous to pharmaceutical requirements |
| Safety assessment | Toxicological profile considering route, quantity, and duration of exposure |
| Benefit/risk evaluation | Demonstrated benefit of substance incorporation vs. risks |
| Interaction assessment | How substance and device interact over the product's intended lifetime |
Classification Rules for Combination Products
Why this matters: Classification determines your conformity assessment route, documentation requirements, and whether you need Notified Body involvement.
MDR Annex VIII establishes classification rules for medical devices. For combination products, classification applies to the device component and directly impacts the regulatory pathway.
Automatic Uplift for Ancillary Substances
Rule 14 of MDR Annex VIII states that devices incorporating a substance that, if used separately, would be considered a medicinal product are automatically classified as:
Class III
When the substance is liable to act on the body with action ancillary to that of the device.
This means device-led DDCs with ancillary substances start at the highest risk class, requiring the most rigorous conformity assessment.
Classification for Medicine-Led DDCs
For integral DDCs where the medicinal product has the principal action, the device classification matters for determining the conformity pathway under Article 117:
Class I (excluding Im, Is, Irsi)
Self-declaration of GSPR compliance acceptable
No Notified Body required for device component
Class Im, Is, Irsi, IIa, IIb, III
Notified Body Opinion required if no EU certificate available
Must confirm full GSPR compliance-partial compliance not acceptable
Conformity Assessment Requirements
Why this matters: The conformity assessment pathway determines what documentation you need in your MAA dossier and how long the device component approval will take.
MDR provides multiple conformity assessment routes depending on device classification. For combination products, Article 117 (which amended Directive 2001/83/EC) specifies what evidence is acceptable.
Article 117 Conformity Evidence Pathways
- 1
EU Declaration of Conformity
Device manufacturer has completed conformity assessment and self-declares compliance
- 2
EU Certificate from Notified Body
NB has certified the device, allowing CE marking (required for higher-risk classes)
- 3
Notified Body Opinion
When no EU certificate exists and device would require NB involvement if standalone
What a Notified Body Opinion Must Confirm
The NB Opinion must confirm full compliance with the relevant GSPRs set out in MDR Annex I. Key points:
- Full compliance required: Partial compliance is not acceptable. CHMP and NCAs don't assess GSPR compliance themselves.
- GSPRs must be listed: The opinion should specify which GSPRs were assessed.
- Applicant can choose any NB: The NB must be designated for the specific type of medical device under MDR.
The Notified Body Role in DDCs
Why this matters: Notified Body capacity is limited, and engagement timelines can extend 12-18 months. Understanding when you need them-and when you don't-is critical for planning.
Notified Bodies play different roles depending on whether your DDC is device-led or medicine-led:
| DDC Type | NB Role | Additional Requirement |
|---|---|---|
| Device-led (ancillary substance) | Full conformity assessment for CE marking | Must obtain EMA/NCA opinion on substance before issuing certificate |
| Medicine-led (higher-risk device) | NB Opinion on GSPR compliance | Opinion included in Module 3.2.R of MAA dossier |
| Medicine-led (Class I device) | Not required | MAH can self-declare GSPR compliance (except Im, Is, Irsi) |
Selecting a Notified Body for DDCs
For combination products, NB selection should consider:
- NANDO designation: NB must be designated for your device type under MDR (not MDD)
- DDC experience: Combination products have unique requirements; experienced NBs reduce review cycles
- Capacity and timeline: Current NB backlogs can extend timelines significantly
- Geographic considerations: EU location requirements for certain activities
For detailed NB selection guidance, see our Notified Body Selection Guide.
Labeling Requirements Under MDR
Why this matters: Labeling errors can delay approvals and require costly re-packaging. Understanding what's required-and what's prohibited-prevents surprises.
MDR Annex I, Chapter III specifies labeling requirements for medical devices. For combination products, the approach depends on whether the device has its own CE mark:
Medicine-Led DDCs with CE-Marked Device
When the device part has its own CE mark (e.g., pre-existing certified component), the integral product labeling follows medicinal product requirements (QRD templates).
UDI consideration: If a UDI is directly marked on the device, it doesn't need to be removed. However, the UDI should NOT appear on the medicinal product's labeling or outer packaging.
Co-Packaged Products
When a medical device is co-packaged with a medicinal product (but not forming an integral product), the device must have its own CE mark and comply with MDR labeling requirements.
Key difference: Co-packaged devices are regulated separately from the medicinal product.
Post-Market Surveillance Obligations
Why this matters: PMS obligations apply immediately-even during transitional periods. Missing these requirements can result in enforcement action.
MDR significantly strengthened post-market surveillance requirements compared to MDD. For combination products, certain MDR PMS obligations apply regardless of which legislation governs the product:
Immediate Application During Transitional Periods
For co-packaged devices benefiting from MDR transitional provisions (Article 120(3a)-(3e)), the following MDR requirements apply immediately:
- Post-market surveillance obligations
- Market surveillance requirements
- Vigilance reporting
- Registration of economic operators
MAH Responsibilities for Device Changes
For all drug-device combinations, the Marketing Authorization Holder must determine whether changes to the device component impact:
- Delivery of the medicinal product
- Quality of the medicinal product
- Safety of the medicinal product
- Efficacy of the medicinal product
If changes lead to a new or updated EU certificate or NB Opinion, this must be submitted as part of an appropriate post-authorization variation procedure. See our EMA Variations Framework 2026 guide for current requirements.
Transitional Provisions and Deadlines
Why this matters: Transitional provisions are complex and have shifted multiple times. Understanding current deadlines prevents costly missteps.
MDR Article 120 provides transitional provisions for devices certified under MDD that don't yet have MDR certification. For combination products, key dates include:
26 May 2021
MDR fully applicable. Article 117 requirements apply to new MAAs.
31 December 2028
End of extended transitional period for devices up-classified under MDR (Article 120(3c) conditions must be met).
Ongoing
Devices placed on market during transitional periods can remain until end of shelf life.
Article 120(3c) Conditions
For devices that didn't require NB assessment under MDD but now do under MDR, the transitional period until 31 December 2028 applies only if:
- No significant changes to design or intended purpose
- Continued compliance with MDD requirements
- Appropriate QMS in place
- MDR requirements for PMS, vigilance, and economic operator registration are met
A Practical Framework for DDC Manufacturers
Why this matters: This framework translates MDR requirements into actionable steps for your regulatory strategy.
Step 1: Determine Your PMOA
Is the principal mode of action pharmacological or device-based?
- • Use MDCG 2022-5 for borderline cases
- • Consider requesting NCA advice for unclear classifications
- • Document your rationale thoroughly
Step 2: Classify the Device Component
Apply MDR Annex VIII classification rules.
- • Remember Rule 14 automatic uplift for ancillary substances
- • Consider all applicable rules (measuring function, software, etc.)
- • Classification drives your conformity assessment requirements
Step 3: Choose Your Conformity Pathway
For medicine-led DDCs, determine which Article 117 pathway applies:
- • Existing EU certificate → Include in Module 3.2.R
- • Self-declaration (Class I only) → MAH statement acceptable
- • No existing assessment → NB Opinion required (12-18 month timeline)
Step 4: Engage Notified Body Early
If NB involvement is required:
- • Check NANDO database for designated NBs
- • Assess NB capacity and experience with DDCs
- • Factor timeline into your development plan
- • Consider parallel submissions where possible
Step 5: Plan for Lifecycle Management
Build processes to manage device changes post-approval:
- • Define change assessment criteria
- • Establish communication with device manufacturer
- • Map changes to variation categories
- • Monitor transitional provision deadlines
References
- 1. Regulation (EU) 2017/745 on medical devices (MDR)
- 2. Questions & Answers for applicants, marketing authorisation holders and notified bodies regarding medicines used in combination with medical devices (EMA/37991/2019 Rev.6, January 2025)
- 3. MDCG 2022-5: Guidance on borderline between medical devices and medicinal products
- 4. EMA Guideline on quality requirements for drug-device combinations (EMA/CHMP/QWP/BWP/259165/2019)
- 5. Directive 2001/83/EC relating to medicinal products for human use (as amended by Article 117)
Need faster answers? RegulatorySense delivers instant, authoritative guidance with EMA document citations.
FAQ
What is the MDR and how does it apply to drug-device combinations?
What are GSPRs and why do they matter for combination products?
When does a DDC need Notified Body involvement under MDR?
What transitional provisions apply to DDCs under MDR?
Confident in Your MDR Compliance Strategy?
The MDR has dozens of provisions affecting combination products. Two minutes to see if you've covered the critical ones.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Check Your Approach →5 questions · Personalized insights · Free