EMA Regulatory
MDR Clinical Investigations: When You Need Them and How to Get Exemptions
Quick Answer
Clinical investigations under MDR are required when existing clinical data is insufficient to demonstrate safety and performance. However, Article 61(4)-(6) provides four exemption pathways: demonstrating equivalence to an existing device, using well-established technology, relying on existing clinical data, or proving the device design eliminates clinical risks. Most manufacturers aim for exemptions-but the burden of proof is on you.
Your Notified Body just sent the dreaded question: "Please provide justification for not conducting a clinical investigation."
You've spent months compiling clinical literature, equivalent device data, and risk analyses. But deep down, you're not certain your justification will hold. The alternative-a full clinical investigation-means 18+ months of delay and costs that could sink the project.
What follows is the decision framework regulatory teams use to know-before they submit-whether an exemption strategy will survive scrutiny.
What Is a Clinical Investigation Under MDR?
A clinical investigation is a systematic study conducted with human subjects to assess the safety or performance of a medical device.
Under MDR Article 2(45), a clinical investigation is defined as:
"Any systematic investigation involving one or more human subjects, undertaken to assess the safety or performance of a device."
Clinical Investigation vs Clinical Evaluation
Don't confuse these two related but distinct concepts:
Clinical Investigation
(one source of clinical data)
- • Prospective study with human subjects
- • Generates NEW clinical data
- • Requires ethics committee approval
- • Follows strict protocol (CIP)
- • May or may not be required
Clinical Evaluation
(ongoing assessment process)
- • Assessment of ALL clinical data
- • Uses existing data sources
- • Required for ALL devices
- • Continuous throughout lifecycle
- • Documented in Clinical Evaluation Report
When Are Clinical Investigations Required?
Why This Matters: Understanding when clinical investigations are mandatory vs optional is the foundation of your regulatory strategy. Get this wrong early, and you'll either waste resources on unnecessary studies or face rejection at conformity assessment.
MDR Article 61 establishes the general principle: manufacturers must conduct clinical investigations unless exemptions apply.
The Default Position
Clinical investigations are required when clinical data from the clinical evaluation is not sufficient to demonstrate conformity with the relevant General Safety and Performance Requirements (GSPRs).
Mandatory for Implantables and Class III
Article 61(4) is explicit: implantable devices and Class III devices shall be subject to clinical investigations-unless exemptions in Article 61(4)(a)-(c), 61(5), or 61(6) apply.
Risk Class and Clinical Investigation Requirements
| Device Class | Clinical Investigation Required? | Exemption Available? |
|---|---|---|
| Class I | Only if data insufficient | Usually not needed |
| Class IIa | Only if data insufficient | Yes, via Article 61(4)-(6) |
| Class IIb | Often required | Yes, but harder to justify |
| Class III | Default: Yes | Yes, but strict requirements |
| Implantables | Default: Yes | Yes, but strict requirements |
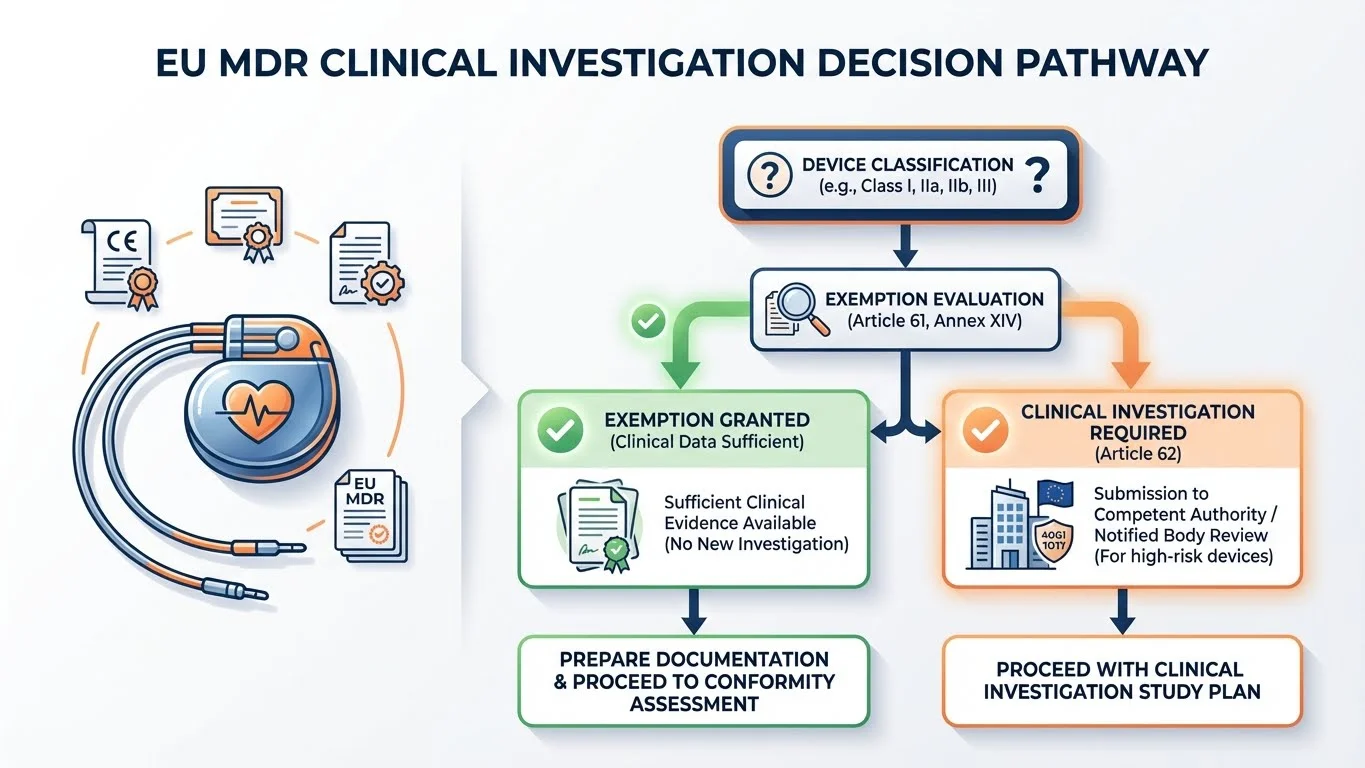
The Four Exemption Pathways Under Article 61
Why This Matters: These exemptions are your alternatives to a full clinical investigation. Understanding exactly what each requires-and what evidence satisfies your Notified Body-is the difference between a 6-month approval and an 18-month delay.
Article 61(4)-(6) provides four pathways to exemption from mandatory clinical investigations for Class III and implantable devices:
Exemption 1: Equivalence with Sufficient Access (Article 61(4)(a))
You can demonstrate equivalence to an existing device AND you have sufficient access to the data relating to that device.
Key requirement: Access means a contract or other arrangement with the original manufacturer giving you rights to the technical documentation, clinical data, and ongoing post-market data.
Exemption 2: Well-Established Technology (Article 61(4)(b))
The device is a modification of a device you already market, AND you can demonstrate equivalence to that device, AND the original device's clinical evaluation is sufficient.
Key requirement: This typically works for iterative improvements to your own devices, not for entering a new market with a competitor's design.
Exemption 3: Existing Clinical Data (Article 61(4)(c))
The device's design and intended purpose are already supported by sufficient clinical data in the scientific literature, making additional clinical investigation unnecessary.
Key requirement: "Sufficient" is judged by your Notified Body. Generic device types with decades of clinical history (e.g., standard hip prostheses) may qualify; novel designs won't.
Exemption 4: Design Eliminates Risks (Article 61(6))
The device's design and construction eliminate or reduce residual risks to acceptable levels, AND existing clinical data (including PMCF) demonstrate acceptable safety and performance.
Key requirement: This applies to devices where inherent safety-by-design arguments are strong. You still need clinical data-just not from a prospective clinical investigation.
Critical Point
Exemptions aren't automatic. You must actively demonstrate that your device meets the exemption criteria. The burden of proof is on the manufacturer, and your Notified Body will scrutinize your justification.
Demonstrating Equivalence: The Three Dimensions
Why This Matters: Equivalence claims are the most common exemption strategy-and the most frequently rejected. Understanding what "equivalent" actually means under MDR is essential.
MDR Annex XIV, Part A defines equivalence across three dimensions-all three must be satisfied:
Technical
- • Similar design
- • Same specifications/properties
- • Similar deployment methods
- • Similar principles of operation
- • Similar conditions of use
Biological
- • Same materials in contact with tissues/body fluids
- • Same surface characteristics
- • Same coatings
- • Same biocompatibility profile
Clinical
- • Same clinical condition
- • Same intended purpose
- • Same site in the body
- • Similar population
- • Similar user profile
The Access Problem
Here's where most equivalence claims fail: under MDR Article 61(5), if you're claiming equivalence to another manufacturer's device, you need:
- A contract with that manufacturer, OR
- Another legal arrangement giving you access to their technical documentation
Without this access, you cannot rely on equivalence to another manufacturer's device-even if your device is technically identical. This is a significant change from the previous Medical Devices Directive (MDD).
Well-Established Technology Exception
MDCG 2020-6 provides guidance on when devices can be considered to use "well-established technology" where clinical investigation may not be needed.
Characteristics of Well-Established Technology
- ✓ Long history of safe clinical use
- ✓ Stable, mature design
- ✓ Well-understood failure modes
- ✓ Extensive clinical literature available
- ✓ Established standards and specifications
- ✓ Predictable performance in clinical use
Important: "Well-established" doesn't mean "common." A device type might be widely used but still have evolving clinical understanding, making it NOT well-established for regulatory purposes.
Examples That May Qualify
- • Standard hip and knee prosthesis designs with 20+ years of clinical data
- • Established pacemaker electrode designs
- • Conventional intraocular lens materials and designs
Note: Novel materials, coatings, or design modifications typically disqualify a device from this exemption.
Clinical Investigation Plan Requirements
Why This Matters: If you do need a clinical investigation, the Clinical Investigation Plan (CIP) is your roadmap-and the Competent Authority's primary review document. Get the CIP wrong, and you'll face validation queries before your investigation even starts.
MDR Annex XV, Chapter II specifies the minimum content for a Clinical Investigation Plan. MDCG 2024-3 provides detailed guidance on preparation.
Essential CIP Elements
- 1. Identification of sponsor and investigators
- 2. Device description and intended purpose
- 3. Rationale for the investigation
- 4. Risk-benefit assessment
- 5. Investigation design and methodology
- 6. Statistical considerations
- 7. Subject selection criteria
- 8. Monitoring procedures
Additional Required Content
- • Clinical endpoints (primary and secondary)
- • Sample size justification
- • Adverse event definitions and reporting
- • Data management procedures
- • Quality assurance measures
- • Informed consent procedures
- • Insurance/indemnification arrangements
- • Publication policy
The Approval Process: Competent Authority and Ethics Committee
Before starting a clinical investigation in the EU, you need approval from both:
- The Competent Authority of each Member State where the investigation will be conducted
- An Ethics Committee in each Member State
Timeline for Competent Authority Review
| Stage | Timeline | Notes |
|---|---|---|
| Validation | 10 days | CA checks completeness of application |
| Assessment | 45 days | CA reviews and may raise questions |
| Clock Stops | Variable | If CA requests additional information |
| Decision | By day 45 | Silence = tacit approval (in most Member States) |
Tacit Approval
If the Competent Authority does not notify the sponsor of refusal within 45 days (plus any clock-stop time), authorization is deemed granted. However, ethics committee approval is still required before starting.
Practical Steps for Regulatory Affairs Professionals
Step 1: Assess Your Clinical Data Position
- 1. Inventory all existing clinical data (literature, PMS data, previous investigations)
- 2. Identify gaps relative to GSPRs and intended purpose
- 3. Determine if equivalence is viable (do you have data access?)
- 4. Evaluate well-established technology arguments
Step 2: Document Your Exemption Justification
- • Prepare detailed equivalence analysis (if claiming equivalence)
- • Compile literature review supporting clinical performance
- • Document risk-benefit analysis for the exemption pathway
- • Engage your Notified Body early-seek informal feedback before submission
Step 3: If Clinical Investigation Required
- 1. Engage clinical operations early-investigation design drives timeline
- 2. Develop CIP with statistical rigor (underpowered studies fail)
- 3. Select Member States strategically (review timelines vary)
- 4. Budget for 12-24 months from CIP finalization to last patient out
Key MDCG Guidance Documents
MDCG 2020-6
Sufficient clinical evidence for legacy devices
MDCG 2021-6
Q&A on clinical investigation requirements
MDCG 2023-7
Clinical investigations exemptions
MDCG 2024-3
Clinical investigation plan preparation
References
- 1. Regulation (EU) 2017/745 on medical devices (MDR), Articles 61-82 and Annex XV
- 2. MDCG 2020-6: Regulation (EU) 2017/745 – Questions and Answers regarding clinical investigation
- 3. MDCG 2021-6: Questions and Answers on clinical investigations
- 4. MDCG 2023-7: Clinical investigations exemptions under Article 61(4)-(6)
- 5. MDCG 2024-3: Guidance on Clinical Investigation Plan
Need faster answers? RegulatorySense delivers instant, authoritative guidance on clinical evidence requirements.
FAQ
Can I avoid a clinical investigation entirely for my Class III device?
What's the difference between a clinical investigation and clinical evaluation?
How long does clinical investigation approval take?
Do I need a clinical investigation if I'm using predicate data from outside the EU?
Need Clarity on Clinical Evidence Requirements?
Clinical investigation decisions shouldn't keep you up at night. Our platform delivers instant, authoritative answers on MDR clinical evidence pathways.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Test Your Regulatory Knowledge → →5 questions · Personalized insights · Free