EMA Regulatory
MDCG Guidance Documents: A Practical Navigation Guide for EU MDR Compliance
Quick Answer
MDCG guidance documents are non-binding interpretations of EU MDR/IVDR issued by the Medical Device Coordination Group. They're organized into six core categories: Clinical Evaluation, Notified Bodies, Post-Market Surveillance, Software/SaMD, UDI/EUDAMED, and Borderline Products. While not legally binding, these documents represent the expected standard for MDR compliance.
You're preparing for a Notified Body audit. The auditor asks about your clinical evaluation approach for a Class IIb implantable device. You cite your internal procedure-but they want to know which MDCG document you followed.
There are over 100 MDCG documents. Some have been revised multiple times. Some overlap. Some contradict earlier positions.
This guide maps the territory so you can find exactly what you need-and know when a document applies to your situation.
What Is the MDCG?
The Medical Device Coordination Group (MDCG) is established under Article 103 of the MDR. It brings together representatives from all EU Member States and is chaired by the European Commission.
The MDCG's primary role is to provide advice and assistance to the Commission and Member States on implementation of the Medical Devices Regulation (EU) 2017/745 and the In Vitro Diagnostic Regulation (EU) 2017/746.
MDCG Subgroups
The MDCG operates through specialized subgroups that focus on specific topics:
- • Borderline and Classification - Product qualification questions
- • Clinical Investigation and Evaluation - CER and clinical study requirements
- • Market Surveillance - PMS and vigilance
- • Notified Bodies - NB designation and oversight
- • Standards - Harmonised standards application
- • UDI and EUDAMED - Database and identification requirements
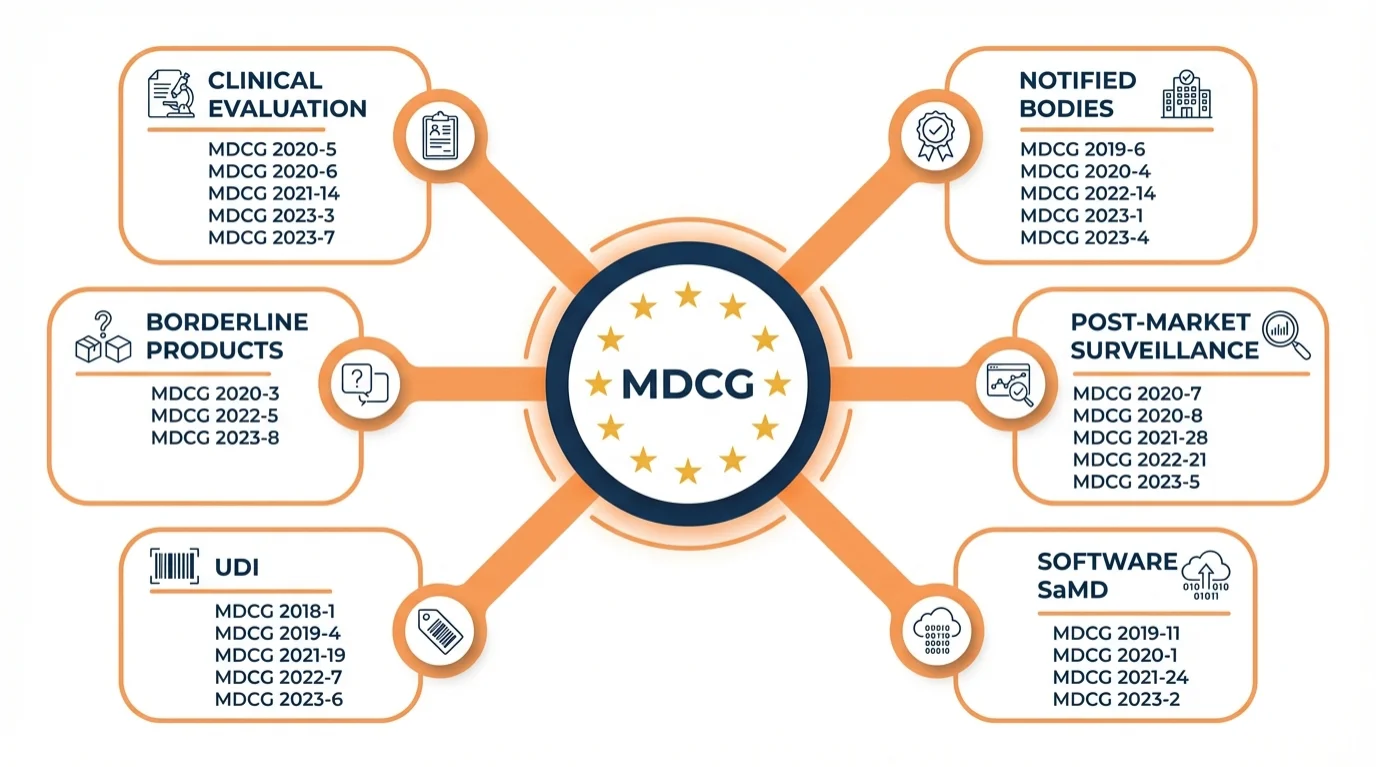
The Six Core Guidance Categories
Why This Matters: Understanding the category structure helps you find relevant documents faster. Instead of scrolling through a long list, you can go directly to the section that addresses your compliance question.
Question
Which MDCG Category Do You Need?
Clinical Evaluation → MDCG 2020-5, 2020-6, 2020-13
Notified Bodies → MDCG 2019-6, 2022-11, 2022-14
Post-Market Surveillance → MDCG 2020-7, 2025-10
Software/SaMD → MDCG 2019-11, 2020-1
UDI/EUDAMED → MDCG 2018-1, 2019-4, 2025-7
Borderline → MDCG 2022-5, Manual on Borderline
Clinical Evaluation Guidance
Clinical evaluation guidance covers how manufacturers should compile and assess clinical evidence to demonstrate conformity with General Safety and Performance Requirements.
| Document | Topic | Key Content |
|---|---|---|
| MDCG 2020-5 | Clinical Evaluation – Equivalence | Criteria for claiming equivalence, contract requirements with equivalent device manufacturer |
| MDCG 2020-6 | Clinical Evaluation – Sufficient Clinical Evidence | What constitutes sufficient evidence, role of post-market data |
| MDCG 2020-13 | Clinical Evaluation – Post-Market Clinical Follow-up | PMCF plan and report requirements, methods for gathering post-market clinical data |
| MDCG 2021-24 | Clinical Evaluation Report Template | Standardized CER structure and content requirements |
| MDCG 2023-7 | Clinical Investigation Q&A | Answers to common questions on clinical investigation conduct |
Key Change from MDD
Under the MDR, claiming equivalence to a competitor's device requires a contract with the competitor giving access to their technical documentation. This is a significant departure from MDD practice and is detailed in MDCG 2020-5.
Notified Body Guidance
These documents address Notified Body designation, surveillance, and the conformity assessment process.
| Document | Topic | Key Content |
|---|---|---|
| MDCG 2019-6 | Questions & Answers for Notified Bodies | Clarifications on NB requirements under MDR Annex VII |
| MDCG 2022-11 Rev.1 | Notified Body Capacity | Recommendations on NB capacity constraints, transparency on timelines |
| MDCG 2022-14 | Transition and NB Availability | Addressing device availability during MDR/IVDR transition |
| MDCG 2022-13 Rev.1 | Joint Assessments of Notified Bodies | Process for NB re-assessments every four years |
| MDCG 2024-12 | CAPA Plans for NB Assessments | Template for corrective and preventive action plans following NB assessment |
Post-Market Surveillance Guidance
PMS guidance covers the manufacturer's obligations to proactively collect and analyze post-market data, as well as vigilance reporting requirements.
MDCG 2020-7
PMS Plan and Report Templates
Provides standardized templates for PMS plans and Periodic Safety Update Reports (PSURs).
MDCG 2025-10
Updated PMS Guidance (December 2025)
Latest guidance on post-market surveillance for medical devices and IVDs. Supersedes earlier versions.
PMS Documentation Requirements
- Post-Market Surveillance Plan per Article 84
- Post-Market Surveillance Report for Class I devices
- Periodic Safety Update Report (PSUR) for Class IIa, IIb, III
- Post-Market Clinical Follow-up (PMCF) Plan and Report
- Trend reporting procedures for incidents
Software and SaMD Guidance
Software guidance addresses qualification (is it a medical device?) and classification of standalone medical device software.
| Document | Topic | Key Content |
|---|---|---|
| MDCG 2019-11 | Software Qualification and Classification | Flowcharts for determining if software is a medical device, classification rules |
| MDCG 2020-1 | Clinical Evaluation of Software | How to apply clinical evaluation requirements to SaMD, including AI/ML considerations |
Software Classification Under MDR Rule 11
MDR Rule 11 significantly changed software classification. Software intended to provide information for diagnostic or therapeutic decisions is now at minimum Class IIa. MDCG 2019-11 provides decision flowcharts to help manufacturers navigate these rules.
UDI and EUDAMED Guidance
These documents cover Unique Device Identification requirements and registration in the European database on medical devices.
| Document | Topic | Key Content |
|---|---|---|
| MDCG 2018-1 | UDI Assignment to Medical Devices | How to assign and use UDI-DI and UDI-PI |
| MDCG 2019-4 | Timelines for UDI Registration | Phased timelines for UDI-DI registration in EUDAMED |
| MDCG 2025-7 Rev.1 | Master UDI-DI Implementation | Updated timelines for contact lenses and spectacle frames (December 2025) |
Borderline Products Guidance
Why This Matters: Borderline guidance helps you determine whether your product is regulated as a medical device at all-or whether it falls under cosmetics, biocides, or pharmaceutical legislation.
MDCG 2022-5
Borderline: Medical Devices vs Medicinal Products
Criteria for distinguishing devices from medicines, including products with ancillary medicinal substances.
Manual on Borderline and Classification
EC Practical Guidance
Extensive manual with product-specific examples covering borderline between devices, cosmetics, biocides, and medicines.
How to Use MDCG Documents in Practice
Step 1: Identify Your Compliance Question
Start with the specific regulatory requirement you need to address. Are you writing a clinical evaluation report? Preparing for a Notified Body audit? Classifying a new software product?
Step 2: Find the Relevant Category
Use the category map above to identify which MDCG subgroup addresses your topic. Check for the most recent version (look for "Rev." suffix).
Step 3: Cross-Reference with MDR Articles
MDCG documents interpret specific MDR articles. Always read the guidance alongside the relevant regulation text to understand both the legal requirement and its practical implementation.
Step 4: Document Your Approach
Reference the MDCG document in your technical documentation. This demonstrates to Notified Bodies and competent authorities that you've followed the agreed interpretation.
Staying Current with MDCG Updates
MDCG documents are published and updated regularly. The official source is the European Commission health portal.
Official MDCG Guidance Source
All MDCG-endorsed documents are published at:
health.ec.europa.eu/medical-devices-sector/new-regulations/guidance-mdcg-endorsed-documents-and-other-guidance_en
Bookmark this page and check it regularly for updates, especially before major submissions or audits.
References
- 1. Regulation (EU) 2017/745 on medical devices (MDR), Article 103 - Medical Device Coordination Group
- 2. MDCG 2020-5: Clinical Evaluation – Equivalence
- 3. MDCG 2020-6: Clinical Evaluation – Sufficient Clinical Evidence
- 4. MDCG 2019-11: Guidance on Qualification and Classification of Software
- 5. MDCG 2025-10: Guidance on Post-Market Surveillance (December 2025)
- 6. European Commission MDCG Guidance Portal
Need faster answers? RegulatorySense delivers instant guidance on MDCG requirements.
FAQ
Are MDCG guidance documents legally binding?
How often are MDCG documents updated?
Where can I find all MDCG guidance documents?
What's the difference between MDCG and CAMD documents?
Find MDCG Requirements Instantly
Stop searching through dozens of guidance documents. Get authoritative answers on MDCG requirements in seconds.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Test Your Regulatory Knowledge → →5 questions · Personalized insights · Free