EMA Regulatory
IVDR Companion Diagnostics: The Complete EMA Consultation Guide
Quick Answer
Under IVDR (EU) 2017/746, companion diagnostics require Notified Body conformity assessment with mandatory scientific opinion from either EMA or a national competent authority. EMA consultation is required when the corresponding medicinal product falls under the centralised procedure. The assessment timeline is 60 days (extendable to 120), based on the draft Summary of Safety and Performance and Instructions for Use.
The Notified Body opinion request comes back with a question she didn't expect: "Please confirm which consultation procedure applies-EMA or national competent authority."
She checks the medicinal product's authorization pathway.
It went through decentralised procedure three years ago. But now there's a line extension under centralised review.

The document that changed everything: IVDR Annex IX Section 5.2, which ties companion diagnostic consultation requirements directly to the medicinal product's authorization route.
What follows is the approach manufacturers use to navigate this intersection of pharmaceutical and device regulation.
What Is a Companion Diagnostic Under IVDR?
Why this matters: The legal definition determines your entire regulatory pathway. Products that seem like companion diagnostics may not meet the IVDR criteria-and vice versa.
Article 2(7) of IVDR defines a companion diagnostic with specific language that carries regulatory weight:
"'Companion diagnostic' means a device which is essential for the safe and effective use of a corresponding medicinal product to:
(a) identify, before and/or during treatment, patients who are most likely to benefit from the corresponding medicinal product; or
(b) identify, before and/or during treatment, patients who are at an increased risk of serious adverse reactions as a result of treatment with the corresponding medicinal product."
The keyword is "essential". A test that provides useful information but isn't required for safe and effective use doesn't meet the CDx definition-even if it's frequently used alongside a therapy.
Common Examples of Companion Diagnostics
Patient Selection CDx
(Identify who will benefit)
- • HER2 testing for trastuzumab
- • EGFR mutation testing for gefitinib
- • BRCA testing for PARP inhibitors
- • PD-L1 testing for checkpoint inhibitors
- • KRAS mutation testing for cetuximab
Safety Monitoring CDx
(Identify who is at risk)
- • HLA-B*5701 for abacavir hypersensitivity
- • TPMT/NUDT15 for thiopurine toxicity
- • UGT1A1 for irinotecan toxicity
- • DPYD for fluoropyrimidine toxicity
- • G6PD for rasburicase contraindication
Classification and Risk-Based Requirements
Why this matters: IVDR classification determines Notified Body involvement and the depth of conformity assessment required.
Companion diagnostics consistently require Notified Body conformity assessment under IVDR-this isn't optional regardless of classification. The regulations recognize that tests essential for treatment decisions carry inherent clinical risk.
Question
What Classification Applies to Your CDx?
Most companion diagnostics fall here-devices where results directly affect patient management decisions for serious conditions
Higher-risk CDx with life-threatening implications if incorrect, or those detecting high-risk pathogens
Most companion diagnostics fall here-devices where results directly affect patient management decisions for serious conditions
Higher-risk CDx with life-threatening implications if incorrect, or those detecting high-risk pathogens
The practical implication: whether your CDx lands in Class C or D, you're looking at Notified Body assessment plus consultation with a medicinal products authority. The difference lies in the depth of scrutiny and whether you need designated reference laboratories.
The EMA Consultation Procedure
Why this matters: This procedure is unique to companion diagnostics. It bridges two regulatory frameworks-pharmaceutical and device-and both must align before your CDx can reach the market.
The consultation procedure requires your Notified Body to seek a scientific opinion on whether your CDx is suitable for use with the corresponding medicinal product. This opinion comes from either EMA or a national competent authority designated under Directive 2001/83/EC.
CDx Consultation Procedure Flow
- 1
Technical Documentation
Manufacturer lodges application with Notified Body including draft SSP and IFU
- 2
NB Initial Assessment
Notified Body assesses device characteristics and IVDR conformity
- 3
Authority Consultation
NB seeks scientific opinion from EMA or national competent authority
- 4
Scientific Opinion
Authority provides opinion within 60 days (extendable to 120)
- 5
Certificate Decision
Notified Body issues EU technical documentation assessment certificate
What the scientific opinion covers:
- Suitability of the CDx for its intended use with the medicinal product
- Adequacy of performance claims relative to clinical decision-making
- Alignment between CDx intended use and medicinal product labeling
- Clinical evidence supporting the CDx-drug relationship
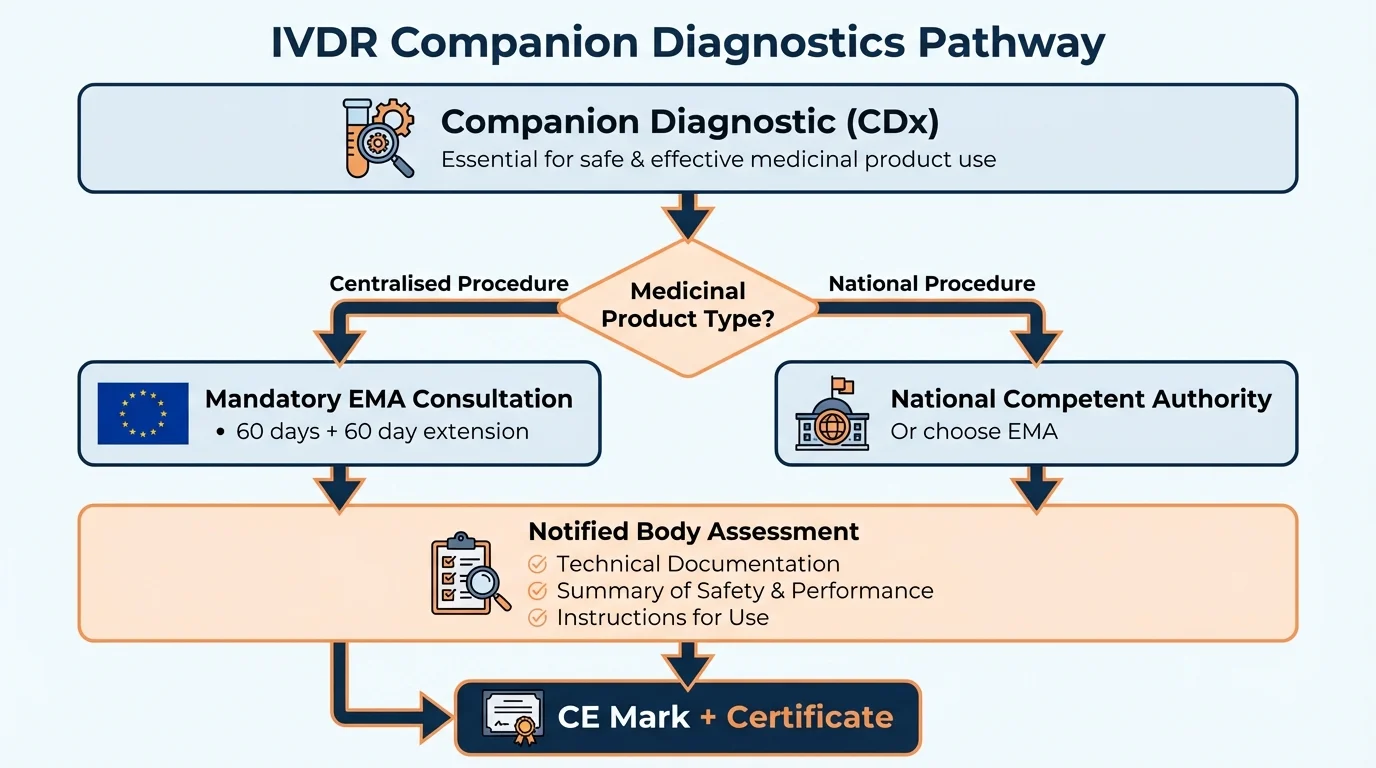
When EMA Consultation Is Mandatory
Why this matters: Getting this wrong doesn't just delay your submission-it can invalidate your conformity assessment entirely.
The IVDR creates a direct link between how the medicinal product was authorized and which authority must be consulted for your companion diagnostic:
CDx Consultation Authority Decision
| Medicinal Product Authorization | EMA Consultation Required | National Authority Option |
|---|---|---|
| Mandatory centralised scope | ✓ Required | ✗ Not permitted |
| Optional centralised (submitted/authorised) | ✓ Required | ✗ Not permitted |
| Decentralised procedure | ○ Optional | ✓ Permitted |
| Mutual recognition | ○ Optional | ✓ Permitted |
| National authorization only | ○ Optional | ✓ Permitted |
The critical point: Mandatory centralised scope includes orphan medicinal products, advanced therapy medicinal products (ATMPs), and products derived from biotechnology processes. If your CDx is intended for any of these, plan for EMA consultation from the start.
Watch for Line Extensions
If a medicinal product originally authorized through national or decentralised procedures later receives a line extension through centralised procedure, your CDx consultation requirements change. The Notified Body must consult EMA for indications covered by the centralised authorization.
Technical Documentation Requirements
Why this matters: The scientific opinion is based on specific documents. Missing or inadequate documentation doesn't just slow the process-it can result in an unfavorable opinion that follows your product.
Two documents form the foundation for the medicinal products authority's scientific opinion:
Summary of Safety and Performance (SSP)
Per IVDR Article 29, the SSP must include:
- • Unique Device Identification (UDI-DI)
- • Intended purpose and indications
- • Summary of performance evaluation
- • Suggested profile of intended users
- • Residual risks and undesirable effects
- • Training required for users
Instructions for Use (IFU)
Per IVDR Annex I Section 20.4.1, the IFU must contain:
- • Intended purpose with therapeutic context
- • Warnings, precautions, and limitations
- • Specimen types and handling requirements
- • Performance characteristics
- • Interpretation guidance for results
- • Medicinal product cross-references
CDx Documentation Completeness Checklist
- Draft SSP includes performance evaluation summary specific to the therapeutic context
- IFU explicitly references the corresponding medicinal product(s)
- Intended use aligns with medicinal product SmPC/label claims
- Clinical evidence demonstrates CDx-drug relationship validity
- Performance claims are appropriate for clinical decision-making
- Analytical validation covers clinically relevant range
- Clinical validation in intended use population documented
Notified Body Assessment Process
Why this matters: The Notified Body is your primary interface, but they're bound by the medicinal products authority's opinion. Understanding this relationship prevents surprises late in the process.
The assessment follows IVDR Annex IX Sections 4.1 to 4.8, with the consultation procedure layered on top. Your Notified Body assesses IVDR conformity while the medicinal products authority evaluates therapeutic suitability.
Timelines You Can Plan Around
| Phase | Timeline | Notes |
|---|---|---|
| Initial NB assessment | Variable | Depends on NB capacity and documentation quality |
| Scientific opinion (standard) | 60 days | From receipt of valid documentation by authority |
| Scientific opinion (extended) | +60 days | On justified grounds, total 120 days |
| Changes requiring supplement | 30 days | For subsequent changes affecting CDx-drug suitability |
Lifecycle Change Management
Any change affecting performance, intended use, or suitability of your CDx in relation to the medicinal product requires notification to your Notified Body. The NB then determines whether a certificate supplement is needed-which triggers another consultation with the medicinal products authority.
Post-Market Surveillance Obligations
Why this matters: CDx post-market surveillance isn't just device surveillance. It requires monitoring the CDx-drug relationship over time, including changes in how the medicinal product is used.
IVDR requires post-market surveillance for all IVDs. For companion diagnostics, this obligation extends to maintaining awareness of how changes in the corresponding medicinal product's use might affect CDx performance expectations.
CDx Post-Market Surveillance Elements
- Performance monitoring specific to therapeutic decision-making context
- Vigilance reporting for incidents affecting diagnosis-treatment link
- Tracking medicinal product label changes that may affect CDx intended use
- Monitoring real-world diagnostic accuracy vs. clinical trial performance
- Post-market clinical follow-up (PMCF) when required
- PSUR preparation per IVDR requirements
FDA vs IVDR: The Regulatory Divergence
Why this matters: If you're pursuing global registration, understanding this divergence affects resource allocation and timeline planning for EU vs. US market access.
In late 2025, FDA announced reclassification of many NGS and NAAT-based oncology companion diagnostics into Class II with special controls. This creates a significant regulatory asymmetry with IVDR.
FDA vs IVDR for Oncology CDx
| Regulatory Element | FDA (Post-2025) | IVDR |
|---|---|---|
| Classification | Class II (reclassified) | Class C (unchanged) |
| Pathway | 510(k) with special controls | NB assessment + authority consultation |
| Agency consultation | Not required | Mandatory for centralised products |
| Timeline | 90 days typical | Variable (NB + 60-120 day opinion) |
| Evidence standards | Harmonized (scientific) | Harmonized (scientific) |
| Regulatory burden | Reduced | Unchanged |
The strategic implication: Scientific standards remain largely harmonized-the same clinical evidence supports both submissions. But regulatory workload is no longer harmonized. US market access is becoming faster for mature CDx technologies, which may influence launch sequence decisions for global programs.
European Commission Reforms (2025-2026)
Why this matters: Proposed reforms directly address CDx-specific burden. Understanding what's coming helps you plan submissions around potential simplifications.
The European Commission's December 2025 proposal (COM(2025) 1023 final) includes specific provisions for companion diagnostics:
These reforms address a core pain point: regulatory duplication. When a CDx is validated for one medicinal product and a manufacturer wants to extend to another with similar clinical context, current rules require full re-consultation. The proposed reforms would limit this to genuinely novel situations.
EUDAMED Requirements from May 2026
From 28 May 2026, manufacturers must register new IVDR devices in EUDAMED before placing them on the EU market. Legacy devices must be registered by 28 November 2026. Certificates issued before these dates must be uploaded by 28 May 2027.
A Practical Framework for CDx Manufacturers
Early Planning Phase
- 1. Map the medicinal product landscape - Identify current and planned authorization pathways for all corresponding products
- 2. Determine consultation authority - EMA mandatory if any centralised procedure products are in scope
- 3. Align intended use language - Your IFU must match SmPC/label claims precisely
- 4. Select Notified Body early - Verify they're designated for IVDR CDx and have EMA consultation experience
Documentation Preparation
- • Build SSP with therapeutic context central, not as an afterthought
- • Cross-reference IFU to medicinal product labeling explicitly
- • Document clinical validation in intended use population, not just analytical performance
- • Prepare for questions about CDx-drug relationship from both NB and authority
- • Assess your documentation readiness before submission
Timeline Planning
- 1. Add buffer for consultation - 60-120 days for scientific opinion, plus NB processing time
- 2. Coordinate with pharma partner - CDx approval often needs to precede or coincide with drug indication approval
- 3. Monitor reform progress - EC proposals may simplify requirements for submissions in late 2026+
- 4. Plan lifecycle changes - Any CDx change affecting the drug relationship restarts the consultation clock
Key Dates and Deadlines
26 May 2022
IVDR came into application
28 May 2026
EUDAMED mandatory for new devices
2026-2027
EC reform implementation (if adopted)
References
- 1. Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR)
- 2. EMA Guidance on procedural aspects for consultation by notified body on companion diagnostics (EMA/747623/2021)
- 3. EMA Questions and answers on implementation of the MDR and IVDR (EMA/37991/2019 Rev.6)
- 4. European Commission Proposal COM(2025) 1023 final - MDR/IVDR Reform
- 5. Team NB Guidance on IVDR Annex IX Section 5.2 (V2, October 2025)
Need faster answers? RegulatorySense delivers instant, authoritative guidance with source citations.
FAQ
What is the definition of a companion diagnostic under IVDR?
When is EMA consultation mandatory for companion diagnostics?
What is the timeline for EMA scientific opinion on CDx?
How do FDA and IVDR approaches to companion diagnostics differ?
Uncertain About Your CDx Consultation Pathway?
EMA or national authority? Centralised or not? One wrong assumption changes your entire timeline. Two minutes to see where you stand.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Check Your Readiness →5 questions · Personalized insights · Free