EMA Regulatory
NB Consultation with EMA: Three Procedures, Three Timelines, One Unified Map
Quick Answer
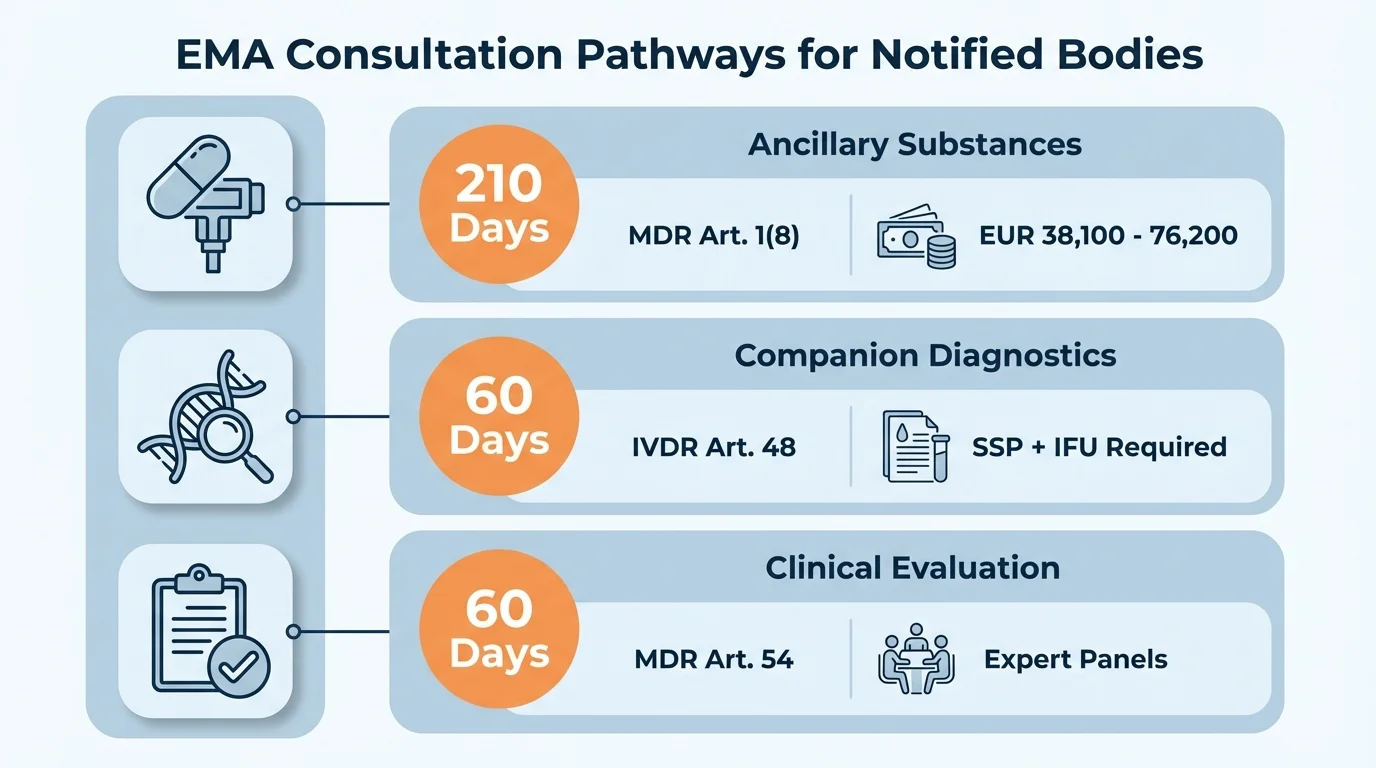
Notified Bodies must consult EMA in three scenarios: devices with ancillary medicinal substances (210-day procedure under MDR Article 1(8)), companion diagnostics (60-day procedure under IVDR Article 48), and certain high-risk devices requiring clinical evaluation consultation with Expert Panels. The NB is always the formal applicant, but the manufacturer prepares the technical dossier and pays the fees. Mandatory vs optional EMA consultation depends on the substance type or the medicinal product's authorization route.
The Notified Body auditor sets down his pen and looks across the table.
"Before we can issue this certificate," he says, "we need to consult EMA."
The room goes quiet. Not because anyone is surprised - they knew this was coming. But because nobody at the table can say with certainty which procedure applies, how long it will take, or what documentation EMA actually expects.

Three different consultation procedures exist under MDR and IVDR - each with its own legal basis, timeline, documentation requirements, and fee structure. And the EMA Q&A documents that explain them are spread across six different sections, multiple revisions, and separate procedural guidance documents that reference each other in circles.
This is the unified map. One reference, three procedures, everything your team needs to know before the NB picks up the phone.
The Three Consultation Types You Need to Know
Why this matters: Each consultation type has fundamentally different requirements, timelines, and consequences. Applying the wrong procedure doesn't just waste time - it can invalidate the conformity assessment entirely.
EMA Consultation Types at a Glance
| Consultation Type | Legal Basis | Timeline | Scope |
|---|---|---|---|
| Ancillary Medicinal Substance | MDR Art. 1(8), Annex IX §5.2 | 210 days (initial) | Quality, safety, usefulness of substance in device |
| Companion Diagnostic | IVDR Art. 48, Annex IX §5.2 | 60 days (initial) | Suitability of CDx in relation to medicinal product |
| Clinical Evaluation (CECP) | MDR Art. 54, Annex IX §5.1 | 60 days | Clinical evaluation assessment of certain Class III/IIb devices |
The first two consultation types involve the CHMP (Committee for Medicinal Products for Human Use) or CAT (Committee for Advanced Therapies). The third - the Clinical Evaluation Consultation Procedure - involves Expert Panels rather than EMA committees, and applies to specific high-risk devices regardless of whether they incorporate medicinal substances.
What makes this confusing for teams is that the same Notified Body might need to initiate different consultation types for different devices in its portfolio, and the procedural requirements for each are documented in entirely separate guidance documents.
Who Consults Whom - And Why It Matters
Why this matters: The roles in this process are counter-intuitive. The manufacturer does most of the work and pays all the fees - but the Notified Body is the one who formally talks to EMA.
Across all three consultation types, the Notified Body is the formal applicant. This isn't just a procedural formality - it means the NB controls the submission timeline, manages the regulatory interaction with EMA, and makes the final certification decision based on EMA's opinion.
Consultation Relationship Map
- 1
Manufacturer Prepares
Device manufacturer prepares technical content - dossier, SSP, IFU, or clinical evaluation report
- 2
NB Submits to EMA
Notified Body submits consultation as formal applicant, including NB's own assessment of suitability/usefulness
- 3
EMA Evaluates
CHMP, CAT, or Expert Panel evaluates and issues scientific opinion or clinical evaluation report
- 4
NB Decides
Notified Body gives due consideration to EMA opinion and makes final certification decision
- 5
Manufacturer Pays
All consultation fees charged directly to the device manufacturer - not the NB
The phrase "due consideration" is deliberately ambiguous. For ancillary blood derivatives, an unfavorable EMA opinion is binding - the NB cannot issue the certificate. For other consultation types, the NB theoretically retains discretion, but deviating from an unfavorable EMA opinion would require extraordinary justification and would likely face challenge during market surveillance.
Ancillary Substance Consultation: The 210-Day Path
Why this matters: This is the longest and most complex of the three consultation procedures - and the one most likely to create project timeline surprises.
When a device incorporates a substance that would be a medicinal product if used separately, and that substance has an action ancillary to the device's function, the quality, safety, and usefulness of that substance must be assessed through consultation. The legal basis is MDR Article 1(8) and Annex IX, Section 5.2.
Question
Is EMA consultation mandatory for your ancillary substance?
EMA consultation MANDATORY - unfavorable opinion blocks certification
EMA consultation MANDATORY - substance falls under centralised procedure
CHOICE between EMA or national competent authority
New MDR consultation still REQUIRED - leverage upcoming variation for MDR-compliant opinion
The 210-day timeline follows the same assessment timetable as a new centralised procedure application, including clock stops for the applicant to respond to questions. Accelerated assessment may be possible when the device addresses a serious disease or uses a known substance from a known source - but justification must be submitted 2-3 months before the planned application.
For a detailed walkthrough of the ancillary substance dossier requirements, variation types, and realistic timelines, see our dedicated guide: Ancillary Substance Consultation: The 210-Day Process That Gates Your CE Mark.
Companion Diagnostic Consultation: The 60-Day Path
Why this matters: The CDx consultation is faster than the ancillary substance path, but its unique complexity lies in the dual dependency between the diagnostic device and the corresponding drug.
Companion diagnostics are defined in IVDR Article 2(7) as devices "essential for the safe and effective use of a corresponding medicinal product." The consultation requirement under Article 48 exists to verify that the diagnostic is genuinely suitable for the therapeutic decision it enables.
EMA consultation is mandatory when the corresponding medicinal product is centrally authorised or falls exclusively within the centralised procedure's scope. For nationally authorised drugs, the NB and manufacturer can choose between EMA and a national authority.
The consultation is based entirely on two documents - the draft Summary of Safety and Performance (SSP) and the draft Instructions for Use (IFU). Unlike the ancillary substance consultation, which evaluates quality and safety comprehensively, the CDx consultation focuses specifically on suitability.
For the complete procedural breakdown including documentation strategies and follow-up consultation triggers, see: CDx Consultation Under IVDR: The 60-Day Procedure That Determines Your Market Access.
Clinical Evaluation Consultation: The Expert Panel Path
Why this matters: This consultation type applies even when your device has no medicinal substance component - it's triggered by device risk class and clinical evaluation questions.
The Clinical Evaluation Consultation Procedure (CECP) under MDR Article 54 is fundamentally different from the other two consultation types. It doesn't involve CHMP or CAT - instead, it routes through Expert Panels established under Article 106 of the MDR.
When CECP Applies
- Class III implantable devices - NB must consult Expert Panel on clinical evaluation assessment report (CEAR)
- Class IIb active devices intended to administer or remove medicinal products
- Certain Class III or IIb devices where the NB identifies specific clinical evaluation concerns
- When a Notified Body has doubts about the adequacy of clinical evidence for a high-risk device
The CECP follows a 60-day timeline and results in a scientific opinion on the Notified Body's clinical evaluation assessment. The Expert Panel may recommend additional clinical investigations or identify gaps in the clinical evidence that the NB must address before certification.
Pro Tip
Unlike the ancillary substance and CDx consultations where the outcome is a single opinion, Expert Panel opinions in the CECP are published. This creates transparency around the clinical evidence basis for high-risk devices, which means your clinical evaluation strategy is, in a sense, public-facing. Plan your CEAR accordingly.
Fee Structure: What Each Consultation Actually Costs
Why this matters: These fees are charged to the manufacturer, not the NB, and they're substantial enough to require budget planning at the project level.
EMA Consultation Fees
| Consultation Scenario | Fee (EUR) | Notes |
|---|---|---|
| New ancillary substance - unknown source | 76,200 | Highest tier - substance + source both new to EMA |
| Known blood derivative - known source | 57,200 | Source previously evaluated by EMA |
| Known substance - known source | 38,100 | Both substance and source previously evaluated |
| Follow-up to initial consultation | 19,100 | Post-approval changes to existing opinion |
| Major amendment (Type II variation) | 43,700 | Significant changes to substance documentation |
| Minor amendment | 7,300 | Administrative or minor quality changes |
Two practical details that are easy to miss: if a device incorporates multiple ancillary substances or blood derivatives, only the highest applicable fee is charged - not a fee for each substance. And manufacturers with registered SME status at EMA's SME office qualify for fee reductions across all consultation types.
EUR 76,200
Maximum EMA consultation fee for a new ancillary substance from an unknown source
This represents the highest fee tier. Known substances from known sources are assessed at EUR 38,100 - less than half the maximum. SME reductions apply where eligible.
FAQ
What are the different types of EMA consultation for Notified Bodies?
Who is the formal applicant in a Notified Body consultation with EMA?
Can a Notified Body choose between EMA and a national authority?
How much do EMA consultation fees cost for medical devices?
What happens after an EMA consultation opinion is issued?
Planning Your First EMA Consultation?
The difference between a 60-day assessment and a 12-month odyssey often comes down to dossier preparation. See where your documentation stands before you file.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Check Your Readiness →5 questions · Personalized insights · Free