EMA Regulatory
CDx Consultation Under IVDR: The 60-Day Procedure That Determines Your Market Access
Quick Answer
Companion diagnostic (CDx) devices under IVDR require a scientific opinion from EMA or a national authority before a Notified Body can issue certification. EMA consultation is mandatory when the corresponding medicinal product is centrally authorised. The initial consultation follows a 60-day timetable, based on the draft Summary of Safety and Performance (SSP) and Instructions for Use (IFU). Follow-up consultations for performance or intended use changes take 30 days.
She's reading the same paragraph for the third time.
Not because it's complicated - because it's circular. The IVDR says the Notified Body must seek a scientific opinion on the "suitability" of the companion diagnostic "in relation to the medicinal product." But what does "suitability" actually mean in practice?
What goes into the dossier? Who decides which authority to consult? And what happens when performance data changes after the initial opinion?

The answers aren't buried - they're scattered. Across Article 48 of the IVDR, Annex IX Section 5.2, Annex X Section 3(k), and separate EMA procedural guidance documents that have been revised multiple times since 2022.
What follows pulls those threads together into a single, navigable framework - the kind of clarity the regulation itself doesn't provide.
What the EMA Q&A Actually Covers
Why this matters: The EMA Q&A sections on companion diagnostics address the procedural gaps that the IVDR itself leaves open - particularly around which authority to consult, what to include in the dossier, and how follow-up consultations work.
The IVDR establishes the legal framework for companion diagnostics in Article 2(7) and sets out the consultation requirement in Article 48. But the regulation's language is deliberately broad - it tells you that a consultation must happen without prescribing exactly how.
The EMA Q&A fills those gaps. It clarifies three things that matter most for teams navigating their first CDx consultation:
What the Q&A Addresses
- Which consultation procedure applies - EMA vs national authority - and when the choice is yours versus when it's mandatory
- What documentation the Notified Body must submit, particularly the role of the SSP and IFU
- How follow-up consultations work when device changes affect suitability
- Timeline expectations for both initial and follow-up assessments
A companion diagnostic under IVDR is defined as a device "essential for the safe and effective use of a corresponding medicinal product." That word - essential - carries significant regulatory weight, because it means the CDx is not optional. Without it, the medicinal product cannot be prescribed safely. And that's precisely why the consultation procedure exists: to verify that the diagnostic is genuinely suitable for the therapeutic decision it enables.
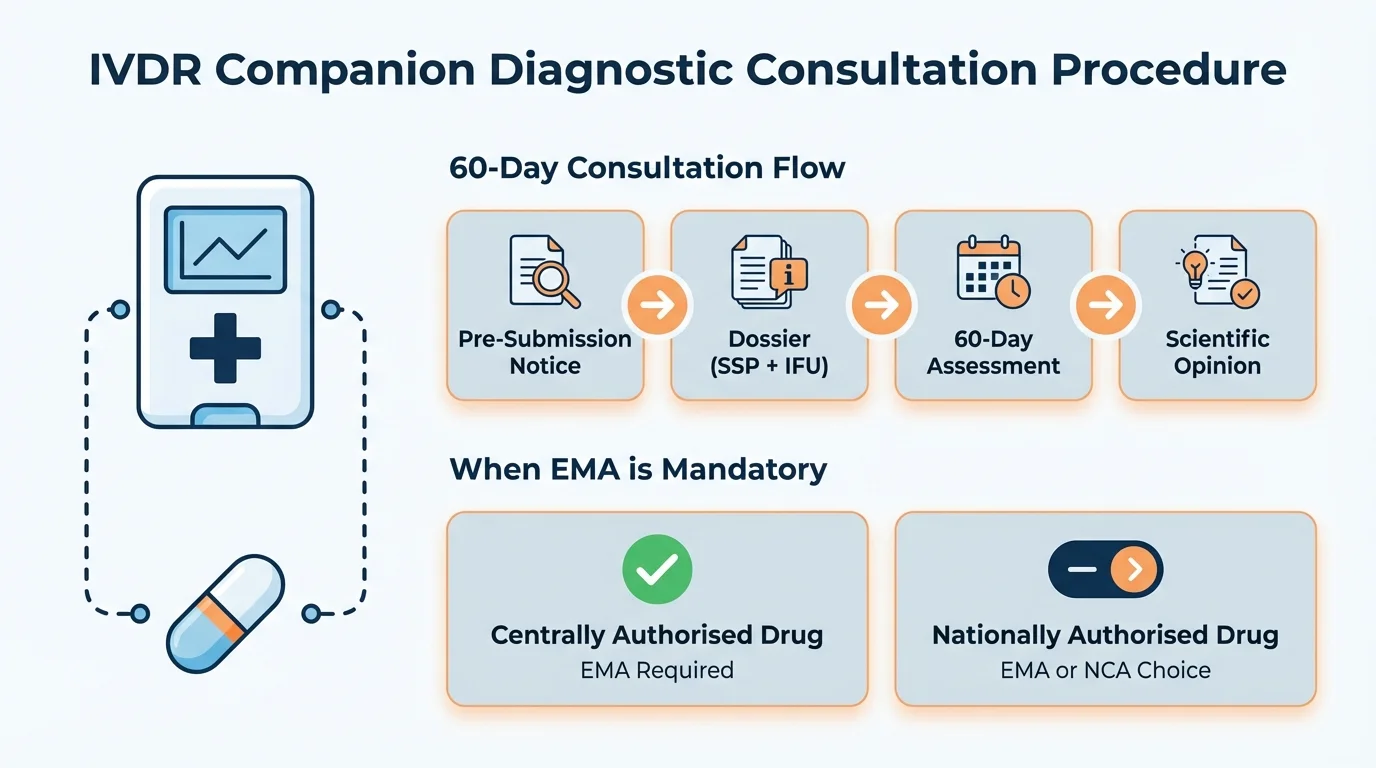
When EMA Consultation Is Required
Why this matters: Getting the authority wrong doesn't just delay your submission - it can invalidate your entire conformity assessment. The decision tree below is the one your team should be using before any consultation is initiated.
The IVDR doesn't automatically route all companion diagnostic consultations through EMA. The determining factor is the regulatory status of the corresponding medicinal product - not the diagnostic device itself.
Question
Which authority must the Notified Body consult for your CDx?
EMA consultation is MANDATORY - no discretion
EMA consultation is MANDATORY - even before MAA submission
Notified Body CHOOSES between EMA or national competent authority
The nuance here is the word "exclusively." Some medicinal products can be authorised either centrally or nationally. If the product can go through a national route, the mandatory EMA requirement doesn't apply - even if the sponsor ultimately chooses the centralised procedure. The trigger is scope, not choice.
Pro Tip
Check the Annex to Regulation (EC) No 726/2004 before initiating consultation. If the corresponding medicinal product appears in the mandatory centralised procedure list, EMA consultation is non-negotiable. If it doesn't, discuss with your Notified Body whether EMA or a national authority offers better alignment with your submission timeline.
The 60-Day Consultation Procedure
Why this matters: Understanding each step - and especially the clock-stop mechanism - prevents the kind of timeline surprises that cascade through your entire development programme.
The consultation procedure for companion diagnostics is shorter than the ancillary substance procedure (60 days vs 210 days), but it carries its own complexities - particularly around who prepares what, and what triggers an extension.
CDx Consultation Procedure
- 1
Pre-Submission Notice
Notified Body notifies EMA at least 1 month before planned submission, specifying scope and submission date
- 2
Dossier Submission
Notified Body submits consultation dossier including draft SSP and IFU, prepared in collaboration with the manufacturer
- 3
Rapporteur Appointment
CHMP or CAT appoints rapporteur(s) to lead evaluation of suitability in relation to the medicinal product
- 4
60-Day Assessment
Committee evaluates analytical and clinical performance data against the medicinal product's therapeutic context
- 5
Questions or Extension
If issues identified, clock stops for response - up to 60 additional days if needed for opinion adoption
- 6
Scientific Opinion
EMA issues opinion on suitability of CDx in relation to the medicinal product - Notified Body must give due consideration
The applicant for the consultation is the Notified Body - not the device manufacturer. This is a critical distinction because it means the Notified Body controls the submission timeline and manages the regulatory relationship with EMA. The manufacturer provides the technical content, but the formal interaction runs through the NB.
That said, EMA's procedural guidance recommends close collaboration between the NB and manufacturer throughout. The consultation dossier should reflect both the NB's conformity assessment perspective and the manufacturer's technical expertise. Misalignment between the two is one of the most common reasons for questions during the assessment period.
What You Need to Submit
Why this matters: The consultation is based entirely on two documents - the SSP and IFU. If these don't contain sufficient performance data, EMA will issue questions that can double your timeline.
Unlike the ancillary substance consultation, which requires a comprehensive dossier covering quality, safety, and usefulness, the CDx consultation focuses specifically on suitability - whether the diagnostic is fit for the therapeutic decision it supports.
CDx Consultation Documentation
| Document | Content Required | Key Focus Areas |
|---|---|---|
| Summary of Safety and Performance (SSP) | Performance evaluation summary, intended purpose, clinical evidence | Analytical sensitivity/specificity, clinical concordance with reference methods |
| Instructions for Use (IFU) | Intended purpose, warnings, precautions, limitations of use | How results guide treatment decisions, specimen requirements, interfering substances |
| NB Verification Report | Notified Body's assessment of suitability rationale | Why this CDx is appropriate for this medicinal product's therapeutic context |
The EMA guidance specifically calls out that "a sufficient level of information regarding the analytical and clinical performance" must appear in both the SSP and IFU. This isn't a vague request - it means EMA expects to see actual performance data, not just references to studies that exist elsewhere.
Pro Tip
Include key performance characteristics directly in the SSP and IFU rather than relying on cross-references to the performance evaluation report. EMA reviewers assess suitability based on what's in these two documents. If critical data requires navigation to separate annexes, expect questions - and questions mean timeline extensions.
Follow-Up Consultations and Changes
Why this matters: Your CDx will evolve - new biomarkers, updated cut-off values, expanded indications for the corresponding drug. Each change triggers a regulatory decision that can either be a 30-day follow-up or a full 60-day re-consultation.
Once an initial consultation opinion is issued, ongoing obligations don't end. Article 48 of the IVDR requires that when changes are made to a CDx that affect its performance, intended use, or suitability in relation to the medicinal product, the manufacturer must inform the Notified Body. The NB then determines whether a follow-up consultation is needed.
Question
What type of follow-up is required when your CDx changes?
30-day follow-up consultation - supplement to existing certificate
New 60-day initial consultation - fresh assessment from scratch
The practical implication is significant: a change to cut-off values for an existing biomarker might qualify as a 30-day follow-up, while adding an entirely new biomarker to the CDx panel would likely trigger a new initial consultation. The Notified Body makes this determination, but EMA's procedural guidance recommends advance notice - ideally at least one month before the planned follow-up submission - so that appropriate planning can occur on both sides.
There's also a post-consultation information flow obligation. If EMA obtains new information about the corresponding medicinal product that could affect the CDx's suitability assessment, EMA is obligated to advise the Notified Body. The NB must then consider this information when reviewing the conformity assessment - potentially triggering a new consultation even without changes to the CDx itself.
Timelines and Fees That Nobody Maps
Why this matters: Your project plan needs real numbers - not regulatory aspirations. The table below shows what to actually budget for.
CDx Consultation Timelines
| Procedure | Regulatory | Realistic |
|---|---|---|
| Initial consultation | 60 days (+60 extension) | 3-5 months total |
| Follow-up | 30 days | 6-8 weeks |
| Re-consultation | 60 days (+60 extension) | 3-5 months |
| Pre-submission prep | 1 month recommended | Plan early |
Fees for CDx consultations follow the same framework established in Council Regulation (EC) No 297/95. The fee is charged to the device manufacturer, not the Notified Body, and SME reductions are available for qualifying manufacturers registered with EMA's SME office.
What most teams underestimate isn't the fee itself - it's the indirect cost. The consultation period happens while your medicinal product development is ongoing, and misalignment between the CDx consultation timeline and the drug's regulatory milestones can create a dependency bottleneck. If your CDx opinion isn't ready when the MAA assessors need it, the entire submission can stall.
FAQ
When is EMA consultation mandatory for companion diagnostics?
How long does the EMA companion diagnostic consultation take?
What documentation is required for a CDx consultation with EMA?
What happens if EMA issues an unfavorable opinion on a companion diagnostic?
Who pays the fees for companion diagnostic consultation with EMA?
Confident Your CDx Dossier Meets EMA Expectations?
One missing performance study or incomplete SSP can trigger a 60-day extension. Two minutes to see where your documentation stands.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Assess Your Readiness →5 questions · Personalized insights · Free