EMA Regulatory
EU MDR Conformity Assessment Procedures: Which Route for Your Device?
Quick Answer
Your device class determines your conformity assessment route. Class IIa and IIb devices can choose between Annex IX (Quality Management System route) or Annex XI (Type Examination + Production QA route). Class III devices must use Annex IX with full product scrutiny. Both routes require Notified Body involvement and lead to CE marking.
You're in a meeting. Someone asks: "Which conformity assessment procedure do we need for this device?"
You pause. You know it depends on device classification. You think Annex IX is the standard route, but you've heard colleagues mention Annex XI for certain devices. The MDR lists multiple pathways, and the consequences of choosing wrong are significant - months of delay, rejected submissions, wasted Notified Body fees.
This guide gives you the decision framework the regulation doesn't spell out clearly.
The Decision Framework: Class Determines Route
Why this matters: Your device class isn't just a regulatory label - it determines your entire conformity assessment pathway, Notified Body involvement level, and documentation requirements.
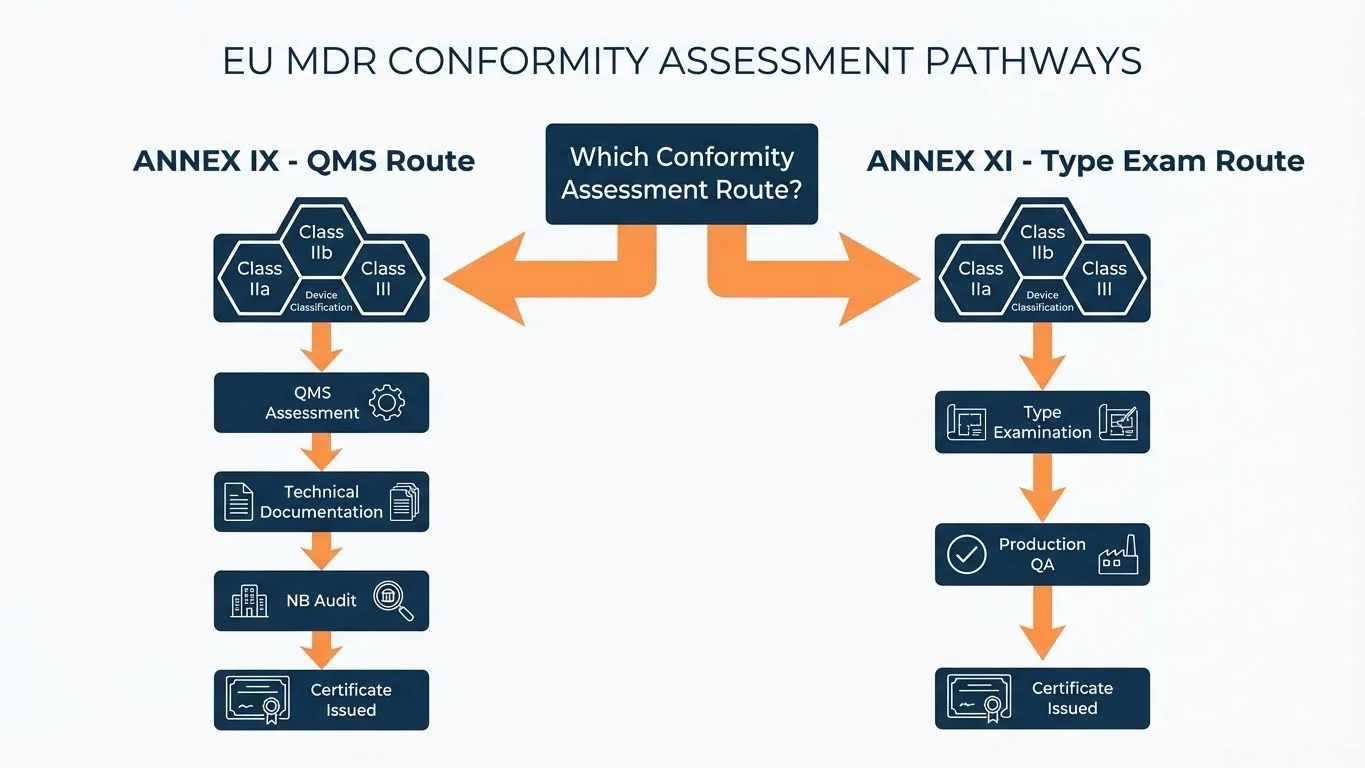
The MDR offers manufacturers choices, but those choices narrow based on device risk classification. Here's the decision tree that actually determines your route.
Conformity Assessment Route Decision
Question
What is your device classification?
Self-assessment (Annex IV) - No Notified Body required for most Class I devices
Choose: Annex IX (QMS Route) OR Annex XI (Type Exam Route)
Choose: Annex IX (QMS + sampling) OR Annex XI (Type Exam + Production QA)
Annex IX with full product scrutiny (Chapter II + III) - Most rigorous pathway
Self-assessment (Annex IV) - No Notified Body required for most Class I devices
Choose: Annex IX (QMS Route) OR Annex XI (Type Exam Route)
Choose: Annex IX (QMS + sampling) OR Annex XI (Type Exam + Production QA)
Annex IX with full product scrutiny (Chapter II + III) - Most rigorous pathway
Most manufacturers default to Annex IX because it's the "standard" route mentioned first in MDR Article 52. But Annex XI exists for good reason - it offers flexibility for manufacturers with product families or frequent design variations.
Pro Tip
Class IIb manufacturers increasingly choose Annex IX over Annex XI. Why? Annex IX allows the Notified Body to sample technical documentation across your product portfolio, while Annex XI requires full technical review for each Type Examination application. If you have multiple device variants, Annex IX scales better.
Annex IX: Quality Management System Route
Why this matters: Annex IX is the most common route for a reason - it's designed for manufacturers with established quality systems and multiple device types. Understanding its two chapters prevents submission errors.
Annex IX has two chapters, and which one applies depends on your device class and risk profile.
Annex IX: Chapter II vs Chapter III
| Aspect | Chapter II (Full QMS) | Chapter II + III (QMS + Scrutiny) |
|---|---|---|
| Applies to | Class IIa, IIb | Class IIb (high-risk), Class III, Class III implantables |
| QMS Assessment | Full quality system audit | Full quality system audit |
| Technical Documentation | Available for review on request | Representative sample reviewed by NB |
| Product Scrutiny | Not required | Required - NB examines design dossiers |
| Certificate | QMS Certificate (Annex IX) | QMS Certificate with product specifics |
| Typical Timeline | 3-6 months | 6-12 months |
Here's what "representative sampling" actually means in Chapter III: The Notified Body doesn't review every device variant. They select devices that represent your technology, risk profile, and design approaches. If you have 50 catheter variants, they might review 5-8 representative designs.
Annex IX Documentation Requirements
- Quality Management System documentation (EN ISO 13485 compliant)
- Technical documentation for all devices (Annexes II and III)
- Clinical evaluation reports for each device family
- Post-market surveillance plan and PSUR commitments
- Risk management files (ISO 14971)
- Declaration of Conformity template
- Label and IFU samples
Annex XI: Type Examination + Production QA Route
Why this matters: Annex XI splits conformity assessment into two parts - product approval first (Type Examination), then production quality approval. This matters when you have design variations or want faster market access for new product versions.
Annex XI works differently. Instead of assessing your entire QMS upfront, the Notified Body first examines a specific device type (Type Examination - Annex X), then verifies your production process can consistently manufacture that approved type.
After Type Examination, you choose one of two production verification routes:
Annex XI Production Routes
| Route | Annex XI Part A: Production QA | Annex XI Part B: Product Verification |
|---|---|---|
| What's assessed | Quality management system for production only | Each product batch or individual devices |
| Best for | High-volume consistent production | Low-volume or custom devices |
| NB involvement | Ongoing production surveillance | Batch-by-batch testing and verification |
| Flexibility | Changes require NB notification | Each batch verified independently |
| Cost structure | Annual surveillance fees | Per-batch verification fees |
Most manufacturers choose Part A (Production QA) because Part B's batch-by-batch verification becomes expensive at scale. But Part B makes sense for truly custom devices or very low production volumes where ongoing surveillance isn't cost-effective.
Pro Tip
If you anticipate frequent design modifications, Annex XI can be faster for getting variants to market. Each Type Examination addition/supplement can cover design changes without triggering full QMS reassessment. With Annex IX, design changes trigger technical documentation sampling reviews.
What the Notified Body Actually Assesses
Why this matters: Knowing what assessors actually look for during audits prevents the most common certification delays. These aren't theoretical requirements - these are the questions your assessment team will ask.
Notified Body assessments aren't abstract. They follow MDR Annex VII requirements and focus on specific evidence. Here's what they're actually checking.
Notified Body Assessment Focus Areas
- QMS implementation evidence (not just procedures - actual records proving the QMS is operational)
- Design control records showing systematic development process
- Risk management integration (not standalone - must link to design, verification, clinical evaluation)
- Clinical evaluation depth and recency (references must be current, search strategy documented)
- Post-market surveillance system (not just a plan - evidence of ongoing data collection and analysis)
- Supplier qualification and control (especially critical for implantables)
- Complaint handling and trend analysis
- Internal audit effectiveness (findings must drive actual improvements)
The most common misconception: manufacturers think Notified Body audits check whether you have the required documents. They actually check whether those documents prove you're consistently applying the processes they describe.
Assessment reports focus on objective evidence. "We have a procedure for X" isn't sufficient - assessors want to see records showing that procedure was followed for actual devices currently on the market.
Common Mistakes That Delay Certification
After analyzing conformity assessment submissions, three mistakes appear repeatedly and cost manufacturers months of delay.
Mistake #1: Incomplete Technical Documentation Before NB Application
Many manufacturers apply for Notified Body assessment before their technical documentation is complete. The logic: "We'll finish it during the assessment process."
Reality: Notified Bodies can't start meaningful assessment without complete technical files. Gaps discovered during assessment trigger major findings, require corrective actions, and extend timelines by 3-6 months. Submit only when Annexes II and III are genuinely complete.
Mistake #2: Misunderstanding "Representative Sampling" for Class IIb/III
Manufacturers with large product portfolios sometimes assume the Notified Body will sample 1-2 devices for technical review.
Reality: MDCG 2019-13 guidance requires sampling plans that genuinely represent your product range. For diverse portfolios, expect 15-25% of devices to be sampled for detailed technical review. If your portfolio has 40 device variants, prepare for 6-10 to undergo full technical documentation assessment.
Mistake #3: Treating Annexes as Separate Requirements
Some teams treat Annex IX (QMS), Annex II (Technical Documentation), and Annex III (Technical Documentation for specific devices) as independent workstreams prepared by different departments.
Reality: Notified Bodies assess integration. They check whether your QMS procedures actually generate the technical documentation described in Annex II, and whether your technical files reference QMS records. Disconnects between these elements trigger major findings about system effectiveness.
Pro Tip
Before starting your conformity assessment application, conduct an internal 'readiness audit' against MDCG 2022-13 (NB designation guidance). This document describes what Notified Bodies must verify. If your internal audit finds gaps, fix them before engaging the NB - it's faster and cheaper than addressing findings during formal assessment.
FAQ
Can a Class IIb device use Annex XI instead of Annex IX?
What is the difference between Annex IX Chapter II and Chapter III?
How long does Notified Body assessment take?
Do I need a new certificate if I change manufacturers?
Confused About Which Conformity Assessment Route to Choose?
Get instant answers to your MDR conformity assessment questions. Our platform delivers authoritative guidance on Annex IX vs Annex XI, Notified Body requirements, and class-specific pathways.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Explore MDR Guidance →5 questions · Personalized insights · Free