EMA Regulatory
Classification Rules for Drug-Device Combinations: The PMOA Decision Framework
Quick Answer
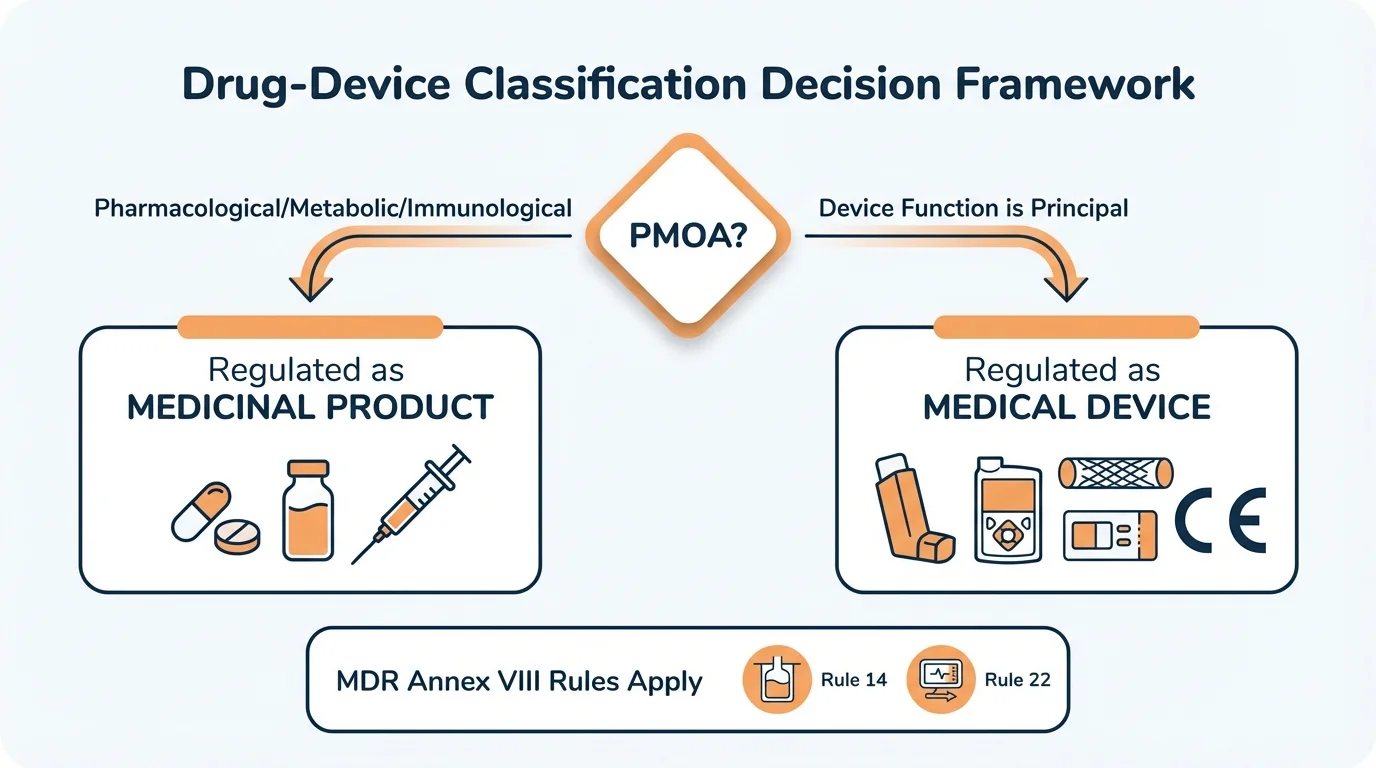
Drug-device combination classification hinges on the Principal Mode of Action (PMOA). If the pharmacological, metabolic, or immunological action is principal, the product falls under pharmaceutical legislation (Directive 2001/83/EC or Regulation 726/2004). If the device function is principal, MDR applies and Rule 14 of Annex VIII classifies devices with ancillary medicinal substances as Class III. The wrong classification means the wrong regulatory framework-and potential rejection of your entire dossier.
The audit finding arrived on a Thursday afternoon. Three words highlighted in red: "Classification inadequately justified."
His team had spent eighteen months preparing the dossier.
The device incorporated a well-known anticoagulant coating-nothing exotic, nothing novel.

What the auditor found: the PMOA determination was based on assumptions, not documented analysis. Was the anticoagulant action principal or ancillary? The dossier didn't definitively answer.
The framework that follows prevents exactly this scenario-by grounding classification in the regulatory logic that auditors and assessors expect to see.
The Foundation: Principal Mode of Action (PMOA)
Why this matters: PMOA isn't just a technical determination-it's the single decision that routes your product into either the pharmaceutical or device regulatory framework. Everything else follows from this.
The demarcation between MDR and pharmaceutical legislation is built on one question: What is the principal intended action of the product?
Question
What is the Principal Mode of Action?
Product regulated as MEDICINAL PRODUCT under Directive 2001/83/EC or Regulation 726/2004
Product regulated as MEDICAL DEVICE under MDR 2017/745 with CE marking requirement
Product regulated as MEDICINAL PRODUCT under Directive 2001/83/EC or Regulation 726/2004
Product regulated as MEDICAL DEVICE under MDR 2017/745 with CE marking requirement
This isn't an arbitrary distinction. Pharmaceutical legislation exists because pharmacological actions carry specific safety considerations-dose-response relationships, metabolic interactions, immunological reactions-that device regulations weren't designed to address. MDR exists because mechanical and physical actions carry their own safety considerations-biocompatibility, sterility, mechanical failure modes.
When Is Action "Principal" vs "Ancillary"?
The regulations don't define a precise threshold. Instead, the determination requires clinical and scientific judgment:
Principal Action (Pharmaceutical Framework)
- • The therapeutic effect depends primarily on the substance
- • Removing the substance would fundamentally change the product's purpose
- • The device component serves mainly to deliver or contain the substance
- • Clinical benefit is mediated through pharmacological/metabolic/immunological pathways
Ancillary Action (Device Framework)
- • The device's mechanical/physical function is the primary therapeutic purpose
- • The substance supports or enhances device function
- • The device would still serve its purpose (perhaps less effectively) without the substance
- • Clinical benefit is primarily mediated through physical mechanisms
MDR Classification Rules That Apply to DDCs
Why this matters: When the device function is principal, specific MDR Annex VIII rules trigger mandatory Notified Body involvement and scientific consultation with medicinal product authorities.
Rule 14: Devices Incorporating Ancillary Medicinal Substances
Rule 14 of MDR Annex VIII applies when a medical device incorporates, as an integral part, a substance which-if used separately-would be considered a medicinal product. The critical word is "ancillary": the medicinal action supports rather than drives the device's purpose.
Automatic Class III Classification
All devices incorporating ancillary medicinal substances are classified as Class III under Rule 14-regardless of what classification they would otherwise receive. This triggers full Notified Body conformity assessment and mandatory consultation with EMA or a national competent authority for medicinal products.
The consultation requirement exists because Notified Bodies have expertise in device conformity assessment but not in pharmaceutical quality and safety. A scientific opinion from a medicinal products authority ensures the ancillary substance is evaluated appropriately.
Rule 22: Devices with Integrated Diagnostic Functions
Rule 22 addresses devices that include an integrated diagnostic function-for instance, a drug delivery device with a sensor that monitors patient response. While not strictly about drug-device combinations, this rule becomes relevant when combination products include monitoring or feedback mechanisms.
Rule 14 Documentation Requirements
- PMOA determination with scientific justification
- Data demonstrating ancillary nature of medicinal substance
- Quality documentation for the medicinal substance (per pharmaceutical standards)
- Safety data for the substance in the context of device use
- Benefit-risk assessment of incorporating the substance
- Stability data demonstrating substance performance over device shelf life
How Pharmaceutical Legislation Interacts with Device Classification
Why this matters: When PMOA is pharmacological, the product falls under pharmaceutical legislation-but device aspects don't disappear. Understanding this interaction prevents gaps in your regulatory strategy.
MDR Article 117 creates the bridge between frameworks. When a medicinal product incorporates a device component (like a pre-filled syringe), the marketing authorization dossier must address both pharmaceutical and device requirements.
Article 117 Documentation Requirements
- 1
Marketing Authorization Dossier
Submit to competent authority or EMA under pharmaceutical legislation
- 2
Device Documentation
Include Declaration of Conformity, CE certificate, or NB opinion on GSPRs
- 3
Dual Assessment
EMA/NCA evaluates drug aspects; NB/NCA evaluates device GSPRs
- 4
Marketing Authorization
Covers entire integral product including device component
The critical insight: The General Safety and Performance Requirements (GSPRs) of MDR Annex I apply to the device part of integral drug-device combinations, even when the product is regulated primarily as a medicinal product. You don't escape device requirements-they're integrated into the pharmaceutical assessment.
Regulatory Framework by PMOA Determination
| Aspect | PMOA = Pharmacological | PMOA = Device Function |
|---|---|---|
| Primary legislation | Directive 2001/83/EC or Reg. 726/2004 | MDR 2017/745 |
| Authorizing body | EMA or National Competent Authority | Notified Body (CE marking) |
| Device requirements | GSPRs (Annex I) via Article 117 | Full MDR conformity assessment |
| Consultation required | NB opinion on GSPRs for device part | EMA/NCA opinion on medicinal substance |
| Market authorization | Marketing authorization covers integral product | CE certificate + DoC |
Classification Scenarios: Which Rules Apply?
Why this matters: Theory is helpful, but regulatory decisions happen in specific product contexts. These scenarios illustrate how classification logic applies in practice.
Scenario 1: Pre-filled Syringe with Biologic
A monoclonal antibody supplied in a pre-filled glass syringe with automatic needle retraction.
Classification: PMOA is pharmacological (the biologic provides the therapeutic effect). The product is an integral drug-device combination regulated as a medicinal product. Article 117 applies-GSPRs for the syringe must be addressed in the marketing authorization dossier.
Scenario 2: Drug-Eluting Coronary Stent
A metal stent coated with antiproliferative drug to prevent restenosis.
Classification: PMOA is debatable but typically determined to be device function (mechanical scaffolding of the vessel). The drug is ancillary. This is a Class III medical device under Rule 14. Notified Body must consult EMA or NCA for opinion on the drug component.
Scenario 3: Antibiotic-Impregnated Bone Cement
PMMA bone cement containing gentamicin for local infection prophylaxis.
Classification: PMOA is device function (mechanical fixation and load bearing). The antibiotic is ancillary. This is a Class III medical device under Rule 14. The antibiotic quality and safety require NCA/EMA consultation.
Scenario 4: Transdermal Patch with Opioid
A transdermal delivery system for fentanyl pain management.
Classification: PMOA is clearly pharmacological (systemic opioid effect). The patch is the delivery mechanism. This is an integral drug-device combination regulated as a medicinal product. The patch component must meet GSPRs within the MA dossier.
IVDR Considerations for Combination Products
Why this matters: If your combination product includes an IVD component, IVDR adds another regulatory dimension. Understanding this prevents pathway confusion.
IVDR (EU) 2017/746 governs in vitro diagnostic devices. When a combination product includes an IVD element-for instance, a drug with an accompanying diagnostic test-the classification analysis becomes multi-dimensional.
Key IVDR Rule
IVD kits may not include medicinal products. If an IVD is intended to be used with a medicinal product and they are co-packaged, the combination cannot be qualified as an IVD kit. Each product must comply with its corresponding legislation separately.
This means companion diagnostics-IVDs essential for safe and effective use of corresponding medicinal products-remain separate products under IVDR, even when closely linked to specific therapies. The classification frameworks don't merge; they coordinate.
Consequences of Misclassification
Why this matters: Classification errors aren't correctable with minor amendments. They can invalidate entire regulatory strategies and require starting over.
The documents are clear: correct qualification and classification of a product is crucial for applying the appropriate set of requirements. Misclassification doesn't trigger a minor correction-it triggers the wrong framework entirely.
Immediate Consequences
- • Conformity assessment under wrong framework is invalid
- • Marketing authorization application rejected
- • CE certificate cannot be issued
- • Notified Body must decline to complete assessment
Downstream Consequences
- • 12-24 month delays to restart under correct framework
- • Significant financial loss (fees, development costs)
- • Competitive disadvantage in market entry
- • Potential enforcement action if product already on market
Dispute Resolution: If a manufacturer and Notified Body disagree on device classification, the dispute is referred to the national competent authority for medical devices in the Member State where the manufacturer is registered. This provides a formal mechanism-but it's far better to resolve classification questions before submission.
A Practical Classification Framework
Step 1: Document the PMOA Determination
- Analyze: What is the primary therapeutic purpose? How is clinical benefit achieved?
- Document: Scientific rationale with literature references
- Justify: Why the pharmacological action is principal or ancillary
- Prepare: Anticipate auditor/assessor questions on the determination
Step 2: Seek Early Guidance for Borderline Products
- • Request informal input from EMA's Innovative Task Force (ITF) for complex cases
- • Consult national competent authority for formal qualification advice
- • Reference MDCG 2022-5 guidance on borderline products
- • Consider pre-submission meetings with Notified Bodies
- • Validate your classification approach before formal submission
Step 3: Apply the Correct Classification Rules
- If PMOA = Pharmacological: Pharmaceutical legislation pathway with Article 117 device requirements
- If PMOA = Device Function: MDR pathway with Rule 14 for ancillary substances
- Document the rule: Explicitly state which classification rule applies and why
- Plan consultation: EMA/NCA consultation for drug aspects; NB for device aspects
References
- 1. Regulation (EU) 2017/745 on medical devices (MDR), Annex VIII Classification Rules
- 2. Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR)
- 3. Directive 2001/83/EC relating to medicinal products for human use
- 4. EMA Questions and answers on implementation of the MDR and IVDR (EMA/37991/2019 Rev.6)
- 5. MDCG 2022-5 Guidance on borderline between medical devices and medicinal products
Need faster answers? RegulatorySense delivers instant, authoritative guidance with source citations.
FAQ
How does PMOA determine classification for drug-device combinations?
What MDR Annex VIII rules specifically apply to drug-device combinations?
What happens if I misclassify a drug-device combination?
Who determines classification for borderline drug-device products?
Confident in Your DDC Classification?
One wrong call on PMOA changes your entire regulatory pathway. Two minutes to validate your classification approach.
Stop searching through hundreds of PDFs. Get authoritative answers in seconds.
Check Your Approach →5 questions · Personalized insights · Free